EEC sendromu , genellikle kalıtsal nitelik gösteren bir sendrom kümesidir. Bu sendrom kümesinde 3 ana bulgu vardır: (1) Ektodermal displazi bulguları, (2) Istakoz kıskacı parmaklar (ectrodactylia), (3) Yarık dudak ve yarık damak.
Costello sendromu, ektodermal displazi bulguları da içeren, otosomal dominant geçen kalıtsal bir sendromdur. Deri, yüz ve iskelet sistemi bulguları ön plandadır. Spontan gen mutasyonuna bağlı az sayıda olgu vardır. Doğumda iri bebek (makrosomi) olsa da, ileriki aylarda belirgin bir gelişme geriliği saptanır. Kısa bir boyun ve büyük bir kafatası (makrosefali) vardır. İnce-kıvırcık-seyrek erken dökülen saçlar erken yaşlanma (progeria) görümü oluşturur.
Ağız-Yüz-Parmak sendromu , günümüze dek 16 fenotipi belirlenmiş olan bir sendromlar kümesidir. Bilinen fenotiplere yenileri eklenebilir. Ağız-Yüz-Parmak sendromunun OFD arasında en sık ratlanılanı OFP tip I temel bulguları içerir; sıkça rastlanan öteki fenotiplerde, tip I'e eklenen ya da çıkarılan yan bulgular vardır.
Carey-Fineman-Ziter sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Genel bir fiziksel ve motor gelişme geriliği vardır. Kafatası asimetriktir (plagiosefali); mikrosefali ya da makrosefali görülebilir. Boyun incedir. Yayvan bir burun vardır. Göz dış bileşikleri aşağı çekiktir, ptozis saptanır. Möbius sequence olgusu nedeniyle okülomotor sinir felci ve oftalmopleji gelişir. Yüz kasları hipoplazisi ya da felci belirlenir. Oral bölge incelemesinde, Pierre Robin sequence bulguları saptanır. Çene eklemi hareketleri sınırlıdır, ağız tam açılamaz. Yutma güçlüğü ve reflü yakınmaları vardır. Erkek çocuklarında skrotuma inmemiş testisler görülür. Eklemlerde kontraktürler olabilir. Vertebra anomalileri ve skolyoz belirlenir. Psikomotor gelişme geriliği nedeniyle hipotoni ve iskelet kaslarında atrofi (myopati) gelişir.

Cornelia de Lange sendromu , 5 fenotipi olan kalıtsal bir sendromdur. Tüm fenotiplerde benzer bulgular saptanır; farklılık etkilenen genlerdedir.

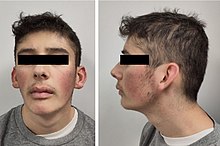
Emanuel sendromu, t(11;22)(q23;q11.2) ayrımı bozukluğu olarak tanımlanan bir kromozom anomalisi sendromudur.
Frontonazal displazi, otosomal resesif yolla aktarılan, alın ve burun bölgelerinin malformasyonlarıyla karakterize kalıtsal bir sendromdur; ALX3 genindeki mutasyonun sonucudur. En önemli bulgu, frontal kemikte ve yüz kemiklerinde orta çizgi yarıklarının bulunmasıdır ile bunun sonucunda ortaya çıkan nazofrontal ensefalosel’dir. Alın derisinde lipoma olabilir. Mikroftalmi saptanır, gözler birbirinden aşırı uzaktadır (hipertelorizm); ptozis, kapak bileşkelerinde anomaliler, katarakt ve epibulbar dermoidler belirlenir. Kulak kepçeleri aşağıdadır, işitme sorunları vardır. Burun kökü aşırı yayvandır; bu alanda saptanan malformasyonların en önemlisi yarıktır. Üstçenede, paranazal sinüslerde ve frontal sinüslerde hipoplazi izlenir. Kalpte Fallot tetralojisi saptanabilir. Pektoral kaslar hipoplazi ya da agenez olabilir. El parmakları kısa ve kıvrıktır. Beyinde Corpus callosum anomalileri saptanır, zeka geriliği vardır. Akromelik frontonazal disostoz fenotipindeki bulgular daha azdır.
Galloway-Mowat sendromu, kalıtsal bir sendromdur. 8 fenotipinin 7'si otosomal resesif yolla aktarılır. Genel gelişme geriliği nedeniyle boy kısadır. Mikrosefali ve küçük bir yüz yapısı saptanır; kafa yanlardan basıktır, alın bombesi yüksektir. Fenotiplerin bir bölümün saptanan çok sayıdaki göz anomalisi nedeniyle görme kusurları belirlenir. Dış kulak ve burun malformasyonları olabilir. Altçene küçüktür (mikrognati), ağız açıklığı malformasyonları ve çukur damak görülebilir. Ağız bulgularının bir bölümü fenotipe özgüdür: mikrodonti, yarık damak, mine hipoplazisi saptanır. Fenotip 4 ve 5'te oral bulgu yoktur.

Joubert sendromu, kalıtsal bir sendromdur; 36 fenotipinden 34'ü otosomal resesif yolla aktarılır, olguların bir bölümü Meckel sendromu ile çakışmaktadır. Joubert sendromunun tüm fenotiplerinde 3 temel bulgu vardır: santral sinir sistemi anomalileri, fizik gelişme geriliği, solunum sorunları. Serebellum (beyincik) vermisi ve beyin sapı hipoplazisi ile beyin radyolojisinde saptanan “azıdişi bulgusu ” santral sinir sistemi anomalilerinin başında gelir. Ataksi ve hipotoni sık görülür. Bazı hastalarda oksipital meningosel ya da meningomyelosel olabilir. Schinzel acrocallosal sendromu, önemli fenotiplerinden biridir.

Kabuki sendromu, iki fenotipi olan bir sendromdur: Kabuki sendromu 1 ve Kabuki sendromu 2 (Kabuk2). Kabuk1, otosomal dominant yolla aktarılan kalıtsal ya da izole olgular biçiminde görülür; Kabuk2 ise, X-kromozomu aracılığıyla dominant yolla (XLD) aktarılır. Her iki fenotipini çok sayıda ortak bulguları vardır; özellikle yüz, iskelet sistemi, gelişme geriliği, parmakizleriyle ilgili bulgular ve zeka geriliği önemlidir.
Mikroftalmi sendromları, 18 fenotipi olan, etkilenen gen türüne göre farklı yollarla -otosomal dominant (AD), otosomal resesif (AR), X-kromozomu dominant (XLD), X-kromozomu resesif (XLR)- aktarılan kalıtsal patolojilerdir. Ortak bulgular yanı sıra farklı sistemlere özgü bulgularla da karşılaşılmaktadır. Ortak bulguların en büyük kümesi gözlerle ilgilidir.
Multipl konjenital anomaliler-hipotoni-epilepsi sendromu, 3 fenotipi olan kalıtsal bir sendromdur. Fenotip 1 (MCAHS1) ve fenotip 3 (MCAHS3) otosomal resesif, tip 2 (MCAHS2) ise x-kromozomu aracılığı ile resesif olarak aktarılır. Birçok enzimin çalışabilmesinde, kompleman sisteminin düzenlenmesinde ve hücrelerarası iletişimin sağlanmasında önemli katkıları olan, fosfogliserid yapısındaki glikosilfosfatidilinositol sentezindeki sorunlarından kökenlidir. Nörolojik belirtiler sendromun fenotiplerindeki temel bulguları oluştururlar; bunlara eklenen çok sayıdaki anomalilerin yeri, sayısı ve gücü farklıdır.
Pallister-Hall sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Hipotalamus'ta hamartoblastoma olarak nitelendirilen oluşumun yanı sıra çok sayıda endokrin sistem anomalileri, açılmamış anüs ve polidaktili bulguları ön plandadır. Hipofiz agenezi/hipoplazisi kökenli hipopituitarizm, adrenal gland hipoplazisi bulguları, hipotiroidizm, endokrin sistem anomalilerinin başlıcalarıdır.

Robinow sendromu, 5 fenotipi olan kalıtsal bir sendromdur; fenotiplerin ikisi otosomal resessif, üçü otosomal dominant yolla aktarılır. Tüm fenotiplerde iskelet sistemi bulguları yoğun olmakla birlikte maksillofasiyal bulguları ile her iki cinste de saptanan genital organ hipoplazileri ortak bulgulardır.

MVA sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. 3 fenotipi vardır; fenotip 1 en sık görülenidir. Anöploidi olarak niterlendirilen kromozom sayısı anomalisinden kökenlidir; kromozom eksikliği (monosomi) ya da fazlalığı (trisomi) vardır. Ancak, bazı hücrelerdeki kromozom sayısının normal olması nedeniyle “mozaisizm” grubu içinde yer alır.
Ritscher-Schinzel sendromu, 2 fenotipi olan kalıtsal bir sendromdur.
Seckel sendromu, 9 fenotipi olan, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Doğumdan önce belirlenen ve yaşam boyu süren, cücelik düzeyinde gelişme geriliği ile kuş kafasını andıran yüz görünümü, mikrosefali ve zeka geriliği saptanır. Alın dardır ve geriye çekilmiş gibidir. Gözler fırlaktır; iki taraflı optik atrofi nedeniyle görme sorunları yaşarlar; gözler çekiktir, kapaklarında yapışıklıklar vardır. Stabismus olabilir. Burun kuş gagasını çağrıştırır. Kulaklar biçimsizdir, kulak memesi yoktur ve işitme güçlüğü belirlenir. Asimetrik bir yüzde, altçene küçüktür ve geridedir (mikroretrognati), damak çukurdur ve yarık saptanır. Dişler küçüktür (mikrodonti) ve eksiktir (hipodonti); bu nedenlerle, çiğneme düzlemi bozuktur (maloklüzyon). Mine hipoplazisi belirgindir.

Toriello-Carey sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Doğum ölçümleri normaldir, ancak zamanla gelişme geriliği bulguları belirir.

Zhu-Tokita-Takenouchi-Kim sendromu (ZTTK sendromu), otosomal dominant yolla aktarılan ya da SON geni mutasyonuyla spontan olarak ortaya çıkan, nörolojik bulguların ön planda olduğu bir sendromdur.

Bohring-Opitz sendromu, otosomal dominant yolla aktarılan, gelişme ve zeka geriliği bulgularının ön planda olduğu, hastaların çoğunun çocukluk yaşlarında kaybedildiği bir sendromdur. C sendromu'nun fenotipi olarak benimsenir.