Cleidocranial dysostosis
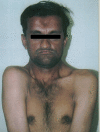
Cleidocranial dysostosis (cleidocranial dysplasia; Scheuthauer-Marie-Sainton sendromu), kemik malformasyonları içeren kalıtsal bir sendromdur; ebeveynlerden birinden otosomal dominant yolla gelir.[1][2] Genetik incelemeler, kemik gelişimiyle ilgili RUNX2 genindeki bir sorunu göstermektedir. 3 tipi vardır; dişlerin etkilenmesi bunlardan yalnızca birinde saptanır. İskelet sistemi etkilenmesinin en tipik bulgusu köprücük kemiklerinde (klavikula;clavicula) görülür. Köprücük kemikleri ya hiç oluşmamıştır (agenezis) ya da gelişmesi yetersizdir (hipoplazi); bu nedenle, hastalar, omuzlarını orta çizgi üzerinde birleştirebilirler. Kafatasındaki bıngıldaklar ve pelvisteki simfizi uzun süre kapanamaz.[1][2][3][4]
Bulgular
- Gelişme geriliği (özellikle yassı kemiklerde)
- Boyun uzun

Cleidocranial dysostosis: Dişlerle ilgili bulgular (persiste süt dişleri ve mine hipoplazisi - Sağ-sol (bilateral) klavikula aplazisi/hipoplazisi
- Kürek kemiği (skapula) hipoplazisi
- Omuzlar dar ve oldukça oynak
- Omurgalarda çökmeler-kaymalar ile kifoz ve lordoz
- Omurilikte kistik oluşumlar (siringomyeli)
- Sternum ve toraks malformasyonları: Kostalar kısadır, toraks dardır
- Sakrum deformasyonları
- Pelvis: pelvisteki simfiz buluşması gerçekleşemez
- Femur boynu kısa, femur başı iridir; femur başı ile ana eksen açısı bozuktır (coxa vara)
- Parmaklarda kalın ve kısa; işaret ve serçe parmaklarının orta bölümündeki kemikler (falanks) kısa
- Kemikler yoğundur (osteoskleroz), kolay kırılırlar
- Brakisefali
- Temporal, frontal ve parietal kemiklerde tümsekleşme
- Foramen magnum geniş
- Doğumda büyük fontaneller
- Kranyum kemiklerinde geciken kaynaşma
- Kafatası kemikleri kalındır
- Mastoid kemik sinüsleri hipoplazisi
- Foramen magnum çok geniştir
- Hipertelorizm
- Burun sırtı yayvan
- Yüz küçük
- Maksiller sinüs hipoplazisi
- Üstçene ve elmacık kemikleri küçüktür
- Çukur damak ve yarık damak
- Altçene uzundur ve öndedir (prognatizm)
- Mandibula yarığı (altçene sağ-sol parçalarının orta çizgide birleşememesi)
- Süt dişlerinin dökülmesinde gecikme (persistans)
- Sürekli dişlerde sürme aksamaları
- Gömük dişler
- Dentigeröz kistler
- Artı (süpernümerer) dişler
- Mesiodens
- Mine hipoplazisi
- Sement hipoplazisi/aplazisi
- Dişlerde kuron ve kök anomalileri
- Maloklüzyon (çiğeneme düzlemi deformasyonu)
Kemik-dışı bulgular
- İşitme sorunları
- Toraks darlığı nedeniyle yenidoğanlarda solunum güçlüğü bulguları
Tedavi
Tüm sendromlarda olduğu gibi, sorunu tümüyle ortadan kaldırmak olanaksızdır; ancak, sorunları hafifletici ve yaşam niteliğini yükseltici önlemler alınabilmektedir. Dişlerle ilgili sorunlarda çene cerrahisi ve ortodonti uzmanlarının, kafatası ve yüz estetiği konularında plastik cerrahi uzmanlarının, onarılması gereken iskelet sistemi malformasyonlarında ise ortopedistlerin katkıları görülmektedir.[3][9]
Kaynakça
- ^ a b c d e Cooper SC, Flaitz CM, Johnston DA, Lee B, Hecht JT. A natural history of cleidocranial dysplasia. American Journal of Medical Genetics, 104:1–6, 2001
- ^ a b c d e Bufalino A, Paranaíba LM, Gouvea AF, et al. Cleidocranial dysplasia: oral features and genetic analysis of 11 patients. Oral Disease, 18:184–190, 2012
- ^ a b c d e Hefti F. Diseases and injuries by site. In “Pediatric Orthopedics in Practice”. 2nd edition, Springer-Verlag, Berlin-heidelberg, 2015
- ^ Mundlos S. Cleidocranial dysplasia: clinical and molecular genetics. Journal of Medical Genetics, 36: 177-182, 1999
- ^ a b Golan I, Baumert U, Hrala BP, Mussig D. Early craniofacial signs of cleidocranial dysplasia. International Journal of Paediatric Dentistry, 14:49–53, 2004
- ^ Cohen MM Jr. Malformations of the Craniofacial Region: Evolutionary, Embryonic, Genetic, and Clinical Perspectives. American Journal of Medical Genetics (Seminars in Medical Genetics), 115:245-268, 2002
- ^ Lubinsky M, Kantaputra PN. Syndromes with supernumerary teeth. American Journal of Medical Genetics, 170A:2611–2616, 2016
- ^ Verma R, Jindal M, Maheshwari S. Familial Cleidocranial Dysplasia. International Journal of Clinical Pediatric Dentistry, 3(1):57-61, 2010
- ^ Park TKN, Vargervik K, Oberoi S. Orthodontic and surgical management of cleidocranial dysplasia. Korean Journal of Orthodontics, 43(5):248-260, 2013










