Tümör (ur; neoplasm; tumor) tanımı önceleri vücuttaki herhangi bir şişlik ya da kitle için kullanılırdı. Sonraları hücrelerin kuralsız ve sınırsız çoğalmaları nedeniyle oluşan kitleler için kullanılmaya başlandı. Yaşamın herhangi bir döneminde organizmanın bir bölümündeki hücreler biyolojik niteliklerini düzenleyici kurallara uyum göstermez ve sınırsız olarak çoğalabilir (otonomi). Bu nitelikleri içeren bir kitleye tümör ya da neoplazm (neoplasm; yeni gelişen kitle) adı verilir. Tümör kitleleri vücudun kendi hücrelerinden yapılıdır.

Hemangioma, kan damarlarının iyi huylu tümörüdür. Olguların çoğu baş-boyun bölgesinde, 1/3'ü karaciğerdedir. Çocuklarda görülen iyi huylu tümörlerin yaklaşık %7'sini oluşturan hemangiomalar, puberteyle birlikte kendiliğinden gerilemekte ve silinmektedir. Cerrahi yöntemlerle çıkarılan oluşumlarda yineleme (residiv) olasılığı vardır.
Paraneoplastik sendrom bir tümör veya tümörün metastazları ile doğrudan ilgili olmayan, yerleşim yerlerinden uzaktaki, ancak tümörün varlığına bağlı olan ve dolayısı ile tümörün çıkarılmasından sonra gerileyebilen belirti ve bulgularıdır.
Dyskeratosis congenita, deri bulguları ön planda olan kalıtsal bir sendromlar kümesidir: otosomal resessif, otosomal dominant ve X kromozomu bağlantılı resessif yollarla aktarılır. 14 fenotipi vardır. X kromozomuyla resessif geçiş gösteren fenotipinde (Zinsser-Cole-Engman sendromu; Hoyeraal-Hreidarsson sendromu) bulgular daha yoğundur. Revesz sendromu, dyskeratosis congenita olgularının kalıtım niteliği otosomal dominant olan fenotipidir.
APECED sendromu, ektodermal displazi bulguları içeren, otosomal dominant ya da otosomal resesif geçen kalıtsal bir sendromdur. İlk belirtiler çocukluk yaşlarında ortaya çıkar. Primer immun yetmezlik sendromlarından biridir.
Beckwith-Wiedemann sendromu, otosomal dominant yolla aktarılan kalıtsal bir aşırı büyüme sendromudur. 4 fenotipi vardır; Konjenital hemihipertrofi (hemihiperplazi) en önemlisidir. Russell-Silver sendromu'nun temel nedenin 11p15 (ICR1) distal kromozomunun yetersiz metilasyonu (hipometilasyon) ise olduğu gösterilmiş; aynı kromozomun hipermetilasyonunun ise Beckwith-Wiedemann sendromuna yol açabileceği ileri sürülmüştür.
Multipl endokrin neoplazi (MEN), endokrin bezlerden kökenli çok sayıda tümörlerinin saptandığı, otosomal dominant yolla aktarılan kalıtsal bir sendrom kümesidir. Endokrin tümörlerin yanı sıra, endokrin nitelik taşımayan organlardan ve dokulardan kökenli tümörler de görülmektedir; endokrin kökenli olsun ya da olmasın, iyi huylu ya da kötü huylu (kanser) tümör özelliklerini taşırlar.

Cowden sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. PTEN hamartoma tümör sendromları kümesi üyelerindendir. Bazı kaynakların listelerinde, PTEN kümesi sendromlarının tümü Cowden sendromu başlığı altında toplanmaktadır.

Proteus sendromu, AKT1 geni somatik mutasyonu ile ortaya çıkar. Bazı kaynaklarda, PTEN hamartoma tümör sendromları kümesi üyelerinden biri olarak nitelendirilir ya da Cowden sendromu başlığı altında toplanan sendrom grubu içinde sayılır. Son yıllarda, aşırı büyüme sendromlarından biri olarak değerlendirilmektedir.

Bannayan-Riley-Ruvalcaba sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. PTEN hamartoma tümör sendromları kümesi üyelerindendir. Bazı kaynakların listelerinde, PTEN kümesi sendromlarının tümü Cowden sendromu başlığı altında toplanmaktadır. Son yıllarda, aşırı büyüme sendromlarından biri olarak değerlendirilmektedir.

Jüvenil hyalin fibromatozis sendromu , otosomal resesif yolla aktarılan kalıtsal bir sendromdur.
Aşırı büyüme sendromları , çocuklarda, doğum öncesi (prenatal) dönemde ya da doğumdan sonra (postnatal) dönemde ortaya çıkabilen, bebeklerin/çocukların vücutlarının tümünde ya da bir bölümünde ortaya çıkan irileşmeyle karakterize olgulardır; aşırı büyümelerin görüldüğü sendromlarda, kilo ve boy artışı, çeşitli anomaliler ile zeka geriliği ve tümör riskinin varlığı en sık rastlanan bulgulardır.
Pallister-Hall sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Hipotalamus'ta hamartoblastoma olarak nitelendirilen oluşumun yanı sıra çok sayıda endokrin sistem anomalileri, açılmamış anüs ve polidaktili bulguları ön plandadır. Hipofiz agenezi/hipoplazisi kökenli hipopituitarizm, adrenal gland hipoplazisi bulguları, hipotiroidizm, endokrin sistem anomalilerinin başlıcalarıdır.

Schinzel-Giedion sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Güçlü bir zeka geriliği ile birlikte garip bir yüz yapısı ve çok sayıda doğumsal anomaliler görülür. Cüceliğe yakın düzeyde boy kısalığına neden olan genel gelişme geriliği vardır.

BOF sendromu, otosomal dominant yolla aktarılan, brankial yarık, göz ve ağız anomalilerinin baskın olduğu, kalıtsal bir sendromdur.

Craniofrontonasal sendrom, X-kromozomu aracılığıyla dominant (XLD) olarak aktarılan kalıtsal bir sendromdur.

Russell-Silver sendromu, genel gelişme geriliğinin neden olduğu boy kısalığı ile yüz ve vücut asimetrisi saptanan; genetik kökeni karmaşık olan, ancak 5 fenotipinden 4’ünün otosomal dominant yolla aktarıldığı belirlenen bir sendromdur. Temel nedenin 11p15 (ICR1) distal kromozomunun yetersiz metilasyonu (hipometilasyon) ise olduğu gösterilmiş; aynı kromozomun hipermetilasyonunun ise Beckwith-Wiedemann sendromuna yol açabileceği ileri sürülmüştür.

Lentigo, melanositleri yerel hiperplazisi sonucu ortaya çıkan, 5–10 mm çapında, oval, koyu ten renginde lekelerdir. Güneş ışınlarının etkisiyle koyulaşmazlar. Bebeklerde ve çocuklarda görece fazladır. Derinin her yerinde, ağız mukozasında, dudaklarda ve gözlerde kapak konjunktivasında görülürler. Ortaya çıkış nedeni bilinmemektedir. Mikroskop incelemelerinde, melanosit hiperplazisinin ve melanin pigmentinin skuamöz epitelin bazal membranında çizgi biçiminde oluştuğu saptanır.
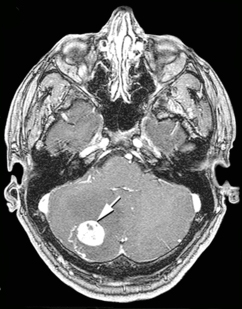
von Hippel-Lindau hastalığı (Hemangioblastoma), otozomal dominant yolla aktarılan kalıtsal bir hastalıktır. Birçok organda kapiller hemangioblastoma olarak adlandırılan tümör vardır. Beyincikte (serebellum) içi sıvıyla dolu kistik bir boşluğu dolduran kapiller hemangioblastoma kitlesi bu hastalıkta karşılaşılan temel bulgudur. Bu yerleşime ek olarak gözde ağ tabakada (retina) ve/veya beyin sapında kapiller hemangioblastoma saptanır. Ayrıca akciğer, karaciğer, böbrek, pankreas, epididim hemangioblastomaları olabilir. Damar tümörü dışında böbrek kanseri, adrenal bezde ya da çevresinde feokromositoma, pankreasta nöroendokrin tümör, kulakta endolenfatik kese tümörü bulunabilir. Böbreklerde, epididimde ve pankreasta kistler vardır. Kan incelemelerinde, hemangioblastomada üretilen eritropoietinin etkisiyle aşırı alyuvar üretimi (polisitemi) saptanır. Ağız mukozasında çok sayıda varis vardır.
Prekanseröz lezyon, bazı hastalıklarda kanser olmayan ancak kanserleşme riski olabilen lezyonlardır. Bu eğilim bazılarında fazla, bazılarında daha azdır. Kanserojen etkilerle bir ya da birden fazla hücrede oluşan DNA zararları ile klinikte tanımlanabilen tümör kitlesinin oluşması arasında sessiz bir dönem (lag period) vardır. Prekanseröz bir lezyonun kansere dönüşmesi (epikarsinogenez) “lag period” ile ilgilidir.
