Seçici serotonin geri alım inhibitörleri majör depresif bozukluk, anksiyete bozuklukları ve diğer psikolojik bozuklukların tedavisinde antidepresan olarak kullanılan bir ilaç grubudur. Yan etkilerinin az olması, etkinlikleri ve tolere edilebilirlikleri nedeniyle sıklıkla depresyon ve diğer birçok psikiyatrik bozukluk için birinci basamak ilaçlar olarak kullanılırlar.
Mutasyon ya da değişinim, bir canlının genomu içindeki DNA ya da RNA diziliminde meydana gelen kalıcı değişmelerdir. Mutasyona sahip bir organizma ise mutant olarak adlandırılır.
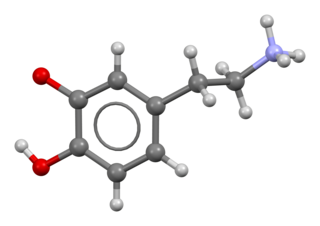
Dopamin, hücrelerde ve canlılarda önemli rol oynayan nöromodülatör bir moleküldür. Çoğu hayvanda ve bazı bitkilerde sentezlenir. Katekolamin ve feniletilamin familyasından olan bir organik bileşiktir. Beyin ve böbreklerde sentezlenen L-DOPA molekülünden bir adet karboksil grubunun çıkarılmasıyla sentezlenen bir amindir. Dopamin, merkezi sinir sisteminde nörotransmiter olarak görev yapar. Nörotransmitterler beynin belirli bölgelerinde sentezlenir, ancak sistemsel olarak birçok bölgeyi etkilerler. Beyin, biri ödül sisteminde önemli bir rol oynayan birkaç farklı dopamin yolağı içerir. Hafıza, hareket, motivasyon, ruh hali ve dikkat süresi dahil olmak üzere birçok vücut fonksiyonunda rol oynar. Genellikle yapılması durumunda sonucunda ödül beklenen eylemler ve aktiviteler, beyindeki dopamin seviyesini artırır. Birçok bağımlılık yapan ilaç dopamin seviyelerini arttırarak çalışır.

Retinitis pigmentosa (RP), halk arasında tavuk karası ve gece körlüğü adlarıyla bilinen ve görme kaybına neden olan genetik bir göz hastalığıdır. Her 4.000 kişide 1'i etkilediği tahmin edilmektedir.

Antidepresanlar, majör depresif bozukluk gibi bazı duygudurum bozukluklarını, bazı anksiyete bozukluklarını ve bazı kronik ağrı durumlarını tedavi etmek için kullanılan ilaçlardır. Antidepresanların yaygın yan etkileri arasında ağız kuruluğu, kilo alımı, baş dönmesi, baş ağrısı, cinsel işlev bozuklukları ve duygusal körelme bulunur. Antidepresanlar, çocuklar, ergenler ve genç yetişkinler tarafından alındığında intihar düşüncesi ve davranışı riskinde artışa neden olabilir. Antidepresanların özellikle ani bir şekilde kesilmeleri sonucunda, antidepresan yoksunluk sendromu ortaya çıkabilir.
Monoamin oksidaz A veya kısaca MAOA, bir insan geni. Bu gen monoamin oksidaz A enzimini kodlar. Bu gendeki bir bozukluk Brunner Sendromuna sebep olur.
Monoamin oksidaz veya MAO, monoaminlerin oksidasyonunu katalizleyen enzimlerdir. Bu enzim Mary Bernheim tarafından ilk olarak karaciğer hücrelerinde tespit edilmiş ve "tyramine oxidase" olarak isimlendirilmiştir. Dopamin, norepinefrin ve serotonin gibi amin nörotransmitterlerini parçalar. Bu protein mitokondri dış zarında bulunur. Monoamin oxidaz A ya da MAOA geni tarafından sentezlenir. Monoamin oksidaz eksikliği Brunner sendromuna sebep olur.
Genetik bozukluk, genlerde ve kromozomlarda görülen anomaliler sonucu ortaya çıkan durumdur. Kanser gibi bazı hastalıklar yaşam sırasında edinilen ve bazı hücrelerde görülen genetik anomaliler nedeniyle olsa da "genetik hastalık" terimi genellikle vücuttaki tüm hücrelerde bulunan ve döllenmeden beri var olan hastalıklar için kullanılır. Bazı genetik bozukluklar, sperm ve yumurtalar gibi üreme hücrelerini oluşturan mayoz bölünme sırasında oluşan kromozom anomalileri nedeniyle ortaya çıkar. Bunlara örnek olarak Down sendromu, Turner sendromu (45X0) ve Klinefelter sendromu sayılabilir. Diğer genetik değişiklikler ebeveynler tarafından tohum hücrelerin oluşturulması sırasında ortaya çıkabilir. Bunlara bir örnek frajil X sendromu ya da Huntington hastalığına neden olabilen üçlü yayılma tekrar mutasyonudur. Hatalı genler ebeveynlerden olduğu gibi alınmış da olabilir. Bu genellikle sağlıklı ama resesif gen taşıyan iki kişinin üremesi ya da hatalı genin dominant olması sonucunda olabilir.
Rett sendromu, yaygın gelişimsel bozukluklardan birisi olarak sınıflandırılan beyinsel gelişim bozukluğudur. Ancak bunun yanlış bir sınıflandırma olduğunu ve benzer şekilde otistik belirtiler gösteren frajil X sendromu, tüberoz skleroz ya da Down sendromunun yaygın gelişimsel bozukluklar olarak sınıflandırılabileceğini önesüren görüşler bulunmaktadır. Bu sendromun belirtileri kolaylıkla otizm ve Angelman sendromunun belirtileriyle karışır. Klinik belirtiler arasında baş büyüme hızının azalması ve bazen mikrosefali, küçük el ve ayaklar bulunur. Stereotipik ve yineleyici el hareketleri de gözlenir. Bilişsel bozukluk ve gerileme döneminde de sosyalleşme sorunları da belirtiler arasında görülür. Okula girdikleri dönemde sosyalleşme genellikle düzelir. Rett sendromu olan kız çocuklar gastrointestinal bozukluklara yakalanmaya yatkındır ve %80’i nöbet geçirir. Hemen hemen hiç sözel becerileri yoktur ve kadınların %50’si yürüyemez. Skolyoz, büyüme eksikliği ve kabızlık çok yaygındır ve sorunlu olabilir.

Otizmin kalıtsallığı, Otizm spektrum bozukluklarının nedenleri arasında en önemli yeri genetik faktörler tutmaktadır. İkizler üzerinde yapılan ilk çalışmalar otizmin kalıtsallığının %90'dan fazla olduğunu, bir başka deyişle genetik faktörlerin otizm vakalarının %90'ından fazlasını açıkladığını göstermiştir. Bu tahminin daha kesinleştirilmesi için ikizler üzerine yeni data ve yapısal genetik modeller gerekmektedir. Tek yumurta ikizlerinden yalnızca biri otistik olduğunda diğerinde genellikle öğrenme ve sosyal bozukluklar görülmektedir. Erişkin kardeşler için ise daha geniş olan otizm fenotipinin bir ya da birkaç özelliğine sahip olma riski %30'dur.
Angelman sendromu ilk olarak 1965 yılında İngiliz doktor Harry Angelman tarafından tanımlanmış nörogenetik bir bozukluktur. Irklarda görülme hızı çok iyi bilinmemekle beraber yaklaşık ensidansın 15,000 ila 30,000 canlı doğumda bir olduğu kabul edilmektedir. Anneden gelen kromozom 15'teki bir bozukluktan kaynaklandığı sanılmaktadır. Hastalığın temel bulguları zeka geriliği, yürüyüş-koordinasyon bozukluğu, konuşma bozukluğu, konvülsiyon ve uygunsuz gülümsemelerdir. Hatta bu sebeple hastalık bazen “mutlu kukla ” sendromu olarak da bilinir.

İnsan genomu Homo sapiens'in genomudur. 23 kromozom çifti üzerinde bulunur, bunlardan 22 çifti otozomal kromozomdur, kalan çift ise cinsiyeti belirler. Haploit insan genomu toplam 3 milyar DNA baz çiftinden biraz fazla uzunluktadır. İnsan Genom Projesi ile elde edilen ökromatik insan genom referans dizisi biyomedikal bilimlerde kullanılmaktadır.
Genetik otostop, bir alelin, olumlu olarak seçilen bir gene bağlanmış olması yüzünden beraberce kalıtıldığı ve böylece alel frekansının ya da görülme sıklığının artığı bir süreçtir. Genlerin bir kromozom üzerindeki birbirine olan yakınlıkları, genlerin, yakınlardaki avantajlı bir gen tarafından maruz kaldığı seçici bir süpürme ile birlikte sürüklenmelerine imkân tanıyabilir. Daha genel olarak, genetik otostop, zararlı mutasyonlara karşı arka planda etki eden seçilim de dahil olmak üzere, birbirine bağlı genler üzerinde etki eden herhangi bir seçilim nedeniyle bir alel frekansında meydana gelen değişikliğe işaret edebilir.
Donohue sendromu, otosomal resesif yolla aktarılan, nadir bir kalıtsal sendromdur. Leprechaunizm ismi, hastalıktan muzdarip olanların çoğu kez elfvari özelliklere sahip olmaları ve olağandan küçük olmalarından gelir (cüce cin sendromu). Ön planda, insülin reseptör defektine bağlı insülin resistansı bulguları saptanır. Pankreasta adacık beta hücrelerinin hiperplazisi nedeniyle kan insülin düzeyi yüksektir (hiperinsülinemi). Spontan abortus ve bebek ölümü riski oldukça yüksektir.

Waardenburg sendromu, en azından bir dereceye kadar doğuştan işitme kaybı ve pigmentasyon eksiklikleri ile karakterize edilen, parlak mavi gözleri, poliosis veya açık ten lekelerini içerebilen bir grup nadir genetik durumdur. Bu temel özellikler, durumun tip 2'sini oluşturur; tip 1, olarak adlandırılan gözlerin iç köşeler arasında daha geniş bir aralık olan telekantus veya distopia kantorum olarak adlandırılan tipi de mevcuttur. Nadir görülen tip 3'te, kollar ve eller de bozuk, kalıcı parmak kontraktürleri veya kaynaşmış parmaklar, tip 4'te ise kişide bağırsak disfonksiyonuna yol açan doğuştan sinir eksikliği olan Hirschsprung hastalığı vardır. Ayrıca, gelişimsel gecikme ve kas tonusu anormallikleri gibi merkezi sinir sistemi semptomlarına neden olabilecek en az iki tip vardır.
XXXY sendromu, bireylerin iki ekstra X kromozomuna sahip olduğu bir cinsiyet kromozomu anöploidi ile karakterize edilen genetik bir durumdur. Çoğu durumda insanlar iki cinsiyet kromozomuna sahiptir: bir X ve bir Y veya iki X kromozomu. İşlevsel bir SRY genine sahip bir Y kromozomunun varlığı, erkekliği belirleyen genlerin ekspresyonuna neden olur. Bu nedenle, XXXY sendromu cinsiyet kimliğinden bağımsız olarak yalnızca biyolojik erkekleri etkiler. XXXY sendromlu erkeklerde ek iki X kromozomu, tipik 46 yerine 48 kromozoma sahip olmalarına neden olur. XXXY sendromu bu nedenle genellikle 48,XXXY olarak adlandırılır. Bilişsel ve davranışsal problemler, taurodontizm ve kısırlık dahil olmak üzere bu sendromla ilişkili çok çeşitli semptomlar vardır. Bu sendrom genellikle ebeveynlerin gametlerinden birinde yeni bir mutasyon yoluyla kalıtılır, çünkü bundan etkilenenler genellikle kısırdır. XXXY'nin her 50.000 erkek doğumdan birini etkilediği tahmin edilmektedir.
Bir örtüşme sendromu, daha yaygın olarak tanınan en az iki bozukluğun özelliklerini paylaşan tıbbi bir durumdur. Örtüşme sendromlarının örnekleri, romatolojide örtüşen bağ dokusu bozuklukları ve kardiyolojide örtüşen genetik bozukluklar gibi birçok tıbbi uzmanlıkta bulunabilir.
MT-TL1 ya da mitokondriyal olarak kodlanmış tRNA lösin 1, insan mitokondri genomundan lösin (UUA/G) için kodlanan bir tRNA genidir.
Kleefstra sendromu, nadir görülen bir genetik bozukluktur. Kromozom 9'un q34 bölgesindeki terminal delesyonlar, çocukluk döneminde hipotoni, belirgin bir yüz görünümü ve gelişimsel engellilik ile ilişkilendirilmiştir. Tipik olarak tanımlanan yüz özellikleri arasında kavisli kaşlar, küçük baş çevresi, orta yüz hipoplazisi, belirgin çene ve dışa doğru çıkıntılı alt dudak bulunur. Bu hastalığa sahip bireylerde sıkça konuşma gecikmeleri gibi konuşma engelleri olabilir. Hastalığın diğer özellikleri arasında: epilepsi, doğumsal ve ürogenital kusurlar, mikrosefali, şişmanlık ve psikiyatrik bozukluklar yer alır. Kromozomal kırılma noktalarının ve önerilen vakalardaki gen diziliminin analizi sonucunda, Dr. Kleefstra ve meslektaşları EHMT1 geninin neden olan gen olduğunu belirledi. Bu gen, histonları değiştirmek için işlev gören histon metiltransferaz proteinini üretmekten sorumludur. Sonuç olarak, histon metiltransferazlar, uygun büyüme ve gelişme için gereken belirli genlerin inaktive edilmesinde önemlidir. Ayrıca, EHMT1'in kodlama dizisindeki bir çerçeve kayması, yanlış anlam ya da anlamsız hatası da bir bireyde bu duruma yol açabilir.
Albinizm-siyah saç teli-hücre göç bozukluğu, bir bireyin fiziksel görünümünü ve fizyolojisini etkileyen durumları tanımlayan terim ve kavramların kısaltmasıdır: (1) A – albinizm, (2) B – siyah saç teli, (3) C – bağırsak nörositlerinin hücre göç bozukluğu ve (4) D – sinirsel tip işitme kaybı. Bu sendrom, endotelin B reseptör geni (EDNRB) mutasyonundan kaynaklanır.