
Yassısolucanlar ya da Platyhelminthes, üç embriyonik tabakadan oluşmuş, bilateral simetrili, çoğunlukla yassı yapılı hayvanlar şubesidir.

Down sendromu, trizomi 21 ya da mongolizm; genetik düzensizlik sonucu insanın 21. kromozom çiftinde fazladan bir kromozom bulunması durumu ve bunun sonucu olarak ortaya çıkan genetik bir bozukluktur. Down sendromu, bireyin 1 yaşından daha uzun süre yaşayabildiği tek otozomal trizomidir.
Otomobil segmentleri, otomobillerin boyut, hacim, fiyat, performans gibi kriterlerle değerlendirilerek oluşturulan sınıflandırmadır.
Diş hekimliğinde, hipodonti edinsel ya da doğumsal diş eksiklikleri olgusu için kullanılan terimlerdendir; anodonti ve oligodonti kavramları da hipodonti başlığı altında yer alan diş eksikliği olgularıdır.
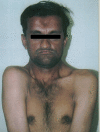
Cleidocranial dysostosis, kemik malformasyonları içeren kalıtsal bir sendromdur; ebeveynlerden birinden otosomal dominant yolla gelir. Genetik incelemeler, kemik gelişimiyle ilgili RUNX2 genindeki bir sorunu göstermektedir. 3 tipi vardır; dişlerin etkilenmesi bunlardan yalnızca birinde saptanır. İskelet sistemi etkilenmesinin en tipik bulgusu köprücük kemiklerinde (klavikula;clavicula) görülür. Köprücük kemikleri ya hiç oluşmamıştır (agenezis) ya da gelişmesi yetersizdir (hipoplazi); bu nedenle, hastalar, omuzlarını orta çizgi üzerinde birleştirebilirler. Kafatasındaki bıngıldaklar ve pelvisteki simfizi uzun süre kapanamaz.
Gorlin-Cohen sendromu , ektodermal displazi bulgularını da içerebilen bir OPD sendromu tipidir. 2 fenotipi vardır:
- Frontometaphyseal dysplasia 1
- Frontometaphyseal dysplasia 2
Mikrognati (micrognathism), altçenenin yetersiz gelişmesi (hipoplazi) niteliğinde bir anomalidir; 1. ve 2. faringeal arklara (brankial yarık) özgü malformasyonların çoğu çene-yüz bölgesindeki mezenkimal dokunun embriyolojik dönemdeki yetersizliğine bağlanmaktadır. Altçene küçüktür ve geridedir; bu olgu, çevredeki kasların hareketlerini sınırlarken bu kaslardan yararlanan dokuların gelişmeleri de kısıtlanır. Kraniyofasiyal anomalileri içeren sendromlarda çok sık görülen bulgulardan biridir. Sendroma-özgü olmayan olguların varlığı da bilinmektedir; örneğin; altçenesi, kafa ile göğüs kafesi arasında sıkışmış olan bir fetüste çene kemiklerinin gelişimi olması gereken düzeye ulaşamamaktadır. Gebelik sorunları, gebelerin alkol kullanması, gestasyonel diabet (gebelik diabeti) gibi çevresel faktörler etkili olabilmektedir (Möbius sendromu). Olguların bir bömümü, gebelerdeki rubella (kızamıkçık) infeksiyonu komplikasyonudur. Yenidoğanların bir bölümünde görülebilen hafif mikrognatiler, altçene gelişiminin tamalanmasıyla silinebilir. Mikrognati'de altçene gövdesi kadar çene eklemini oluşturan yapıların hipoplazisi de önemlidir. Mikrognatilerin bir bölümü kulak sistemi, üstçene ve damak gelişiminin de aksadığı olgularla birlikte görülür.
Kraniyosinostoz, kraniyosinostozis (craniosynostosis), kraniyofasiyal malformasyonların ve maksillofasiyal sendromların önemli bir bölümünde etkileri görülebilen konjenital bir patolojidir. Bu olgudaki temel bulgu kafatası eklemlerinin erken kapanmasıdır; etkilediği anatomik bölgelerde ortaya çıkan malformasyonlar, hangi suturaların ne düzeyde kapanmış olmasıyla orantılıdır. Malformasyonlar genellikle etkilenen eklemin dikey yönünde belirgindir.

Makrosefali , nitelemesi kafatasının aşırı büyüklüğünü tanımlar. Primer makrosefali olgularının büyük bölümü sendroma-özgü bir bulgu olarak ortaya çıkar; bazıları ise başka bir bulgu olmaksızın görülen “ailesel makrosefali”lerdir. Sekonder makrosefali olguları, bir hastalığın temel bulgusu ya da komplikasyonu olarak ortaya çıkarlar ; başka bir bölümü ise, kafatası suturaları kapanmadan önceki dönemlerde geçirilen infeksiyonlar, subdural ya da intraventriküler kanamalar gibi çevresel nedenlere bağlı olabilir. Makrosefali, hidrosefalisi olan çocuklardaki en tipik bulgulardan biridir. Primer ya da sekonder olgularda makrosefalinin neden olduğu nörolojik bulgular saptanabilir.

Kraniyofasiyal yarıklar, kraniyofasiyal malformasyonların en önemlilerinden biridir; baş-boyun ve yüz bölgesinin oluşma ve gelişme aşamalarındaki aksamalar ya da sapmalar sonucu ortaya çıkan yapısal ve işlevsel bozuklukların önemli bir bölümünü oluştururlar. Embriyolojik kökenlerine göre; nöral tüp kökenli anomaliler, 1. ve 2. farengeal ark kökenli malformasyonlar, ektodermal displaziler söz konusudur.
Diastrofik displazi, otosomal resesif yolla aktarılan kalıtsal bir sendromdur; 2 fenotipi vardır. Genel gelişme geriliği saptanır; yenidoğanda kollar ve bacaklarda belirgin biçimde kısadır (cücelik). Kaşlar bölgesinde (glabella) damar tümörü (hemangioma) görülür. Kulak kepçeleri büyüktür, kistik oluşumlar vardır. İşitme sorunları bulunur. Ağız içi incelemede, yarık damak saptanır. Larinks stenozu nedeniyle ses kabadır. Vertebralardaki hipoplaziler nedeniyle kifoskolyoz ve servikal kifoz oluşur. Tüm tubuler kemikler kalın ve kısadır. Diz eklemleri gevşek, kalça eklemleri sorunludur.
Frontonazal displazi, otosomal resesif yolla aktarılan, alın ve burun bölgelerinin malformasyonlarıyla karakterize kalıtsal bir sendromdur; ALX3 genindeki mutasyonun sonucudur. En önemli bulgu, frontal kemikte ve yüz kemiklerinde orta çizgi yarıklarının bulunmasıdır ile bunun sonucunda ortaya çıkan nazofrontal ensefalosel’dir. Alın derisinde lipoma olabilir. Mikroftalmi saptanır, gözler birbirinden aşırı uzaktadır (hipertelorizm); ptozis, kapak bileşkelerinde anomaliler, katarakt ve epibulbar dermoidler belirlenir. Kulak kepçeleri aşağıdadır, işitme sorunları vardır. Burun kökü aşırı yayvandır; bu alanda saptanan malformasyonların en önemlisi yarıktır. Üstçenede, paranazal sinüslerde ve frontal sinüslerde hipoplazi izlenir. Kalpte Fallot tetralojisi saptanabilir. Pektoral kaslar hipoplazi ya da agenez olabilir. El parmakları kısa ve kıvrıktır. Beyinde Corpus callosum anomalileri saptanır, zeka geriliği vardır. Akromelik frontonazal disostoz fenotipindeki bulgular daha azdır.

Bardet-Biedl sendromu, hipofiz ve hipotalamus kökenli endokrin sistem sendromlarından biridir. Kalıtsaldır. 24 fenotipi vardır, bunların yalnızca 3’ü önemlidir ve otosomal dominant ya da otosomal resesif yolla aktarılır.

Konjenital spondiloepifizeal displazi , otosomal resesif yolla aktarılan, kondrodisplazi grubundan, iskelet sistemi malformasyonlarının yoğun olduğu kalıtsal bir sendromdur. Doğumdan önce belirlenen cücelik düzeyinde gelişme geriliği vardır. Cüceliğin nedeni gövde kısalığı, uzun kemiklerin epifizlerindeki malformasyonlar ve yassılaşmış omurgalardır. Boyun kısadır.

BOF sendromu, otosomal dominant yolla aktarılan, brankial yarık, göz ve ağız anomalilerinin baskın olduğu, kalıtsal bir sendromdur.

Rosselli-Gulienetti sendromu, otosomal resesif yolla aktarılan, ender görülen kalıtsal bir sendromdur. Klinik bulgular ektodermal displazi izlenimi verir. Saçlar, kaşlar ve kirpikler seyrektir ve hızla dökülürler. Kulaklar önde ve yelkensi görünümdedir. Ağıziçi incelemelerde, yarık dudak ve yarık damak saptanır. Dişler küçüktür (mikrodonti) ve eksiklikler vardır (hipodonti). Bazı hastalar dişsizdir (anodonti). Parmak derileri yapışıktır (sindaktili). Avuçiçleri ve ayak tabanlarının derisindek keratin tabakası kalındır. Terleme yetersizliği olabilir. Tırnak yapıları bozuktur. Psikomotor gerilik vardır.

Wolf-Hirschhorn sendromu, genel gelişme geriliği, kafa ve yüz malformasyonları ve epileptiform atakların görüldüğü, “4. kromozomun kısa kolunun distal kısmında delesyon (4p-)” olarak bilinen kromozom anomalisi sonucu ortaya çıkan izole olgulardır. Kız hastalar görece sıktır; 1/3’ü iki yaşına ulaşamadan kaybedilir.
BOR sendromu, otosomal dominant aktarılan kalıtsal bir sendromdur.
Otofasiyoservikal sendrom (otofaciocervical sendrom), yüz ve iskelet malformasyonlarının ön planda olduğu, 2 fenotipi saptanan bir sendromdur. Fenotiplerde genler farklıdır; biri otosomal dominant, öteki otosomal resesif yolla aktarılır. BOR sendromu ile aleldir.
Fryns mikroftalmi sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Mikroftalmi sendromları kümesinin elemanıdır.