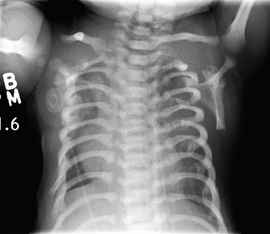
Ellis-van Creveld sendromu, ektodermal displazi bulguları içeren, otosomal resesif geçen kalıtsal bir sendromdur. Asfiksiyan torasik displazi sendromu'nun 22 fenotipinden 2'si Ellis-van Creveld sendromu olarak değerlendirilir.
Costello sendromu, ektodermal displazi bulguları da içeren, otosomal dominant geçen kalıtsal bir sendromdur. Deri, yüz ve iskelet sistemi bulguları ön plandadır. Spontan gen mutasyonuna bağlı az sayıda olgu vardır. Doğumda iri bebek (makrosomi) olsa da, ileriki aylarda belirgin bir gelişme geriliği saptanır. Kısa bir boyun ve büyük bir kafatası (makrosefali) vardır. İnce-kıvırcık-seyrek erken dökülen saçlar erken yaşlanma (progeria) görümü oluşturur.
Kraniyoektodermal displazi (Cranioectodermal dysplasia), ektodermal displazi bulguları da içeren, otosomal resesif geçen kalıtsal bir sendromdur. 4 fenotipi vardır:
- Cranioectodermal dysplasia 1 (Sensenbrenner sendromu; Levin sendromu; arthrodentoosseous dysplasia)
- Cranioectodermal dysplasia 2
- Cranioectodermal dysplasia 3
- Cranioectodermal dysplasia 4
Ağız-Yüz-Parmak sendromu , günümüze dek 16 fenotipi belirlenmiş olan bir sendromlar kümesidir. Bilinen fenotiplere yenileri eklenebilir. Ağız-Yüz-Parmak sendromunun OFD arasında en sık ratlanılanı OFP tip I temel bulguları içerir; sıkça rastlanan öteki fenotiplerde, tip I'e eklenen ya da çıkarılan yan bulgular vardır.
OPD sendromu (otopalatodigital sendrom), X-kromozomu aracılığıyla dominant (XLD) yolla geçen kalıtsal bir sendromdur (Gorlin-Cohen sendromu hariç). Kız çocukları daha hafif etkilenir. Tümü benzer bulgular içeren 5 tipi vardır:
- Otopalatodigital sendrom tip I (OPD tip1)
- Otopalatodigital sendrom tip II (OPD tip2)
- Gorlin-Cohen sendromu (ilgili sayfaya gidiniz)
- Melnick-Needles sendromu (tip 1 ile allelik bağlantısı vardır; ilgili sayfaya gidiniz)
- Terminal osseöz displazi ve pigmentli deri defektleri (çok enderdir, ayrıntı verilmemiştir).
Gorlin-Cohen sendromu , ektodermal displazi bulgularını da içerebilen bir OPD sendromu tipidir. 2 fenotipi vardır:
- Frontometaphyseal dysplasia 1
- Frontometaphyseal dysplasia 2
Beckwith-Wiedemann sendromu, otosomal dominant yolla aktarılan kalıtsal bir aşırı büyüme sendromudur. 4 fenotipi vardır; Konjenital hemihipertrofi (hemihiperplazi) en önemlisidir. Russell-Silver sendromu'nun temel nedenin 11p15 (ICR1) distal kromozomunun yetersiz metilasyonu (hipometilasyon) ise olduğu gösterilmiş; aynı kromozomun hipermetilasyonunun ise Beckwith-Wiedemann sendromuna yol açabileceği ileri sürülmüştür.
Kraniyosinostoz, kraniyosinostozis (craniosynostosis), kraniyofasiyal malformasyonların ve maksillofasiyal sendromların önemli bir bölümünde etkileri görülebilen konjenital bir patolojidir. Bu olgudaki temel bulgu kafatası eklemlerinin erken kapanmasıdır; etkilediği anatomik bölgelerde ortaya çıkan malformasyonlar, hangi suturaların ne düzeyde kapanmış olmasıyla orantılıdır. Malformasyonlar genellikle etkilenen eklemin dikey yönünde belirgindir.

Makrosefali , nitelemesi kafatasının aşırı büyüklüğünü tanımlar. Primer makrosefali olgularının büyük bölümü sendroma-özgü bir bulgu olarak ortaya çıkar; bazıları ise başka bir bulgu olmaksızın görülen “ailesel makrosefali”lerdir. Sekonder makrosefali olguları, bir hastalığın temel bulgusu ya da komplikasyonu olarak ortaya çıkarlar ; başka bir bölümü ise, kafatası suturaları kapanmadan önceki dönemlerde geçirilen infeksiyonlar, subdural ya da intraventriküler kanamalar gibi çevresel nedenlere bağlı olabilir. Makrosefali, hidrosefalisi olan çocuklardaki en tipik bulgulardan biridir. Primer ya da sekonder olgularda makrosefalinin neden olduğu nörolojik bulgular saptanabilir.
Frontonazal displazi, otosomal resesif yolla aktarılan, alın ve burun bölgelerinin malformasyonlarıyla karakterize kalıtsal bir sendromdur; ALX3 genindeki mutasyonun sonucudur. En önemli bulgu, frontal kemikte ve yüz kemiklerinde orta çizgi yarıklarının bulunmasıdır ile bunun sonucunda ortaya çıkan nazofrontal ensefalosel’dir. Alın derisinde lipoma olabilir. Mikroftalmi saptanır, gözler birbirinden aşırı uzaktadır (hipertelorizm); ptozis, kapak bileşkelerinde anomaliler, katarakt ve epibulbar dermoidler belirlenir. Kulak kepçeleri aşağıdadır, işitme sorunları vardır. Burun kökü aşırı yayvandır; bu alanda saptanan malformasyonların en önemlisi yarıktır. Üstçenede, paranazal sinüslerde ve frontal sinüslerde hipoplazi izlenir. Kalpte Fallot tetralojisi saptanabilir. Pektoral kaslar hipoplazi ya da agenez olabilir. El parmakları kısa ve kıvrıktır. Beyinde Corpus callosum anomalileri saptanır, zeka geriliği vardır. Akromelik frontonazal disostoz fenotipindeki bulgular daha azdır.

Joubert sendromu, kalıtsal bir sendromdur; 36 fenotipinden 34'ü otosomal resesif yolla aktarılır, olguların bir bölümü Meckel sendromu ile çakışmaktadır. Joubert sendromunun tüm fenotiplerinde 3 temel bulgu vardır: santral sinir sistemi anomalileri, fizik gelişme geriliği, solunum sorunları. Serebellum (beyincik) vermisi ve beyin sapı hipoplazisi ile beyin radyolojisinde saptanan “azıdişi bulgusu ” santral sinir sistemi anomalilerinin başında gelir. Ataksi ve hipotoni sık görülür. Bazı hastalarda oksipital meningosel ya da meningomyelosel olabilir. Schinzel acrocallosal sendromu, önemli fenotiplerinden biridir.
Mikroftalmi sendromları, 18 fenotipi olan, etkilenen gen türüne göre farklı yollarla -otosomal dominant (AD), otosomal resesif (AR), X-kromozomu dominant (XLD), X-kromozomu resesif (XLR)- aktarılan kalıtsal patolojilerdir. Ortak bulgular yanı sıra farklı sistemlere özgü bulgularla da karşılaşılmaktadır. Ortak bulguların en büyük kümesi gözlerle ilgilidir.

Robinow sendromu, 5 fenotipi olan kalıtsal bir sendromdur; fenotiplerin ikisi otosomal resessif, üçü otosomal dominant yolla aktarılır. Tüm fenotiplerde iskelet sistemi bulguları yoğun olmakla birlikte maksillofasiyal bulguları ile her iki cinste de saptanan genital organ hipoplazileri ortak bulgulardır.

Potter sequence olgularında belirlenen önemli neden oligohidramnios bulgusudur.

Meckel-Gruber sendromu, çok sayıda genin etkilenmesinden kaynaklanan, bu nedenle 15 fenotipi olan, otosomal resesif yolla aktarılan, siliyopatiler grubundan, kalıtsal bir sendromdur. Kistik böbrekler, beyin anomalileri ve ensefalosel, polidaktili ve karaciğer anomalileri başlıca bulgulardır. En sık görülen fenotip Meckel sendromu 1, öteki fenotiplerdeki bulguların çok büyük bir bölümünü içerir. Bazı fenotipleri Joubert sendromuyla çakışan nitelikleri içermektedir.
Saldino-Noonan sendromu, otosomal resesif yolla aktarılan asfiksiyan torasik displazi sendromu’nun 22 fenotipinin 3.sü olan kalıtsal bir sendromdur. Asfiksiyan torasik displazi sendromu’nun ortak bulgularına ek olarak, dilde hamartomalar, klavikulaların bisiklet gidonu biçiminde olması, cinsel organlarda malformasyonlar, enkondral ossifikasyon bozuklukları, tibia yokluğu ve skolyoz vardır.
Majewski sendromu, otosomal resesif yolla aktarılan asfiksiyan torasik displazi sendromu’nun 22 fenotipinin 6.sı olan kalıtsal bir sendromdur. Asfiksiyan torasik displazi sendromu'ndaki ortak bulguların büyük bölümü Majewski sendromu'nda da saptanmaktadır. Dudak yarığının orta çizgide olması, epiglot ve larinks malformasyonları ile klavikula ve skapula anomalileri farklı olan bulgulardır.
Mainzer-Saldino sendromu, otosomal resesif yolla aktarılan asfiksiyan torasik displazi sendromu'nun 22 fenotipinin 9.su olan kalıtsal bir sendromdur. Asfiksiyan torasik displazi sendromu'ndaki ortak bulguların büyük bölümü Mainzer-Saldino sendromu'nda da saptanmaktadır. Üçgenimsi kafatası yapısı (trigonosefali), ön-arka düzlemde uzun kafatası (skafosefali), görme sorunlarına neden olan retinitis pigmentosa, ağız açıklığının büyük olması (makrostomi), damak ve dudak yarıklarının bulunmaması, dişlerin yerleştiği kemikte düzensizlikler görece sık rastlanan farklılıklardır.

Beemer-Langer sendromu, otosomal resesif yolla aktarılan asfiksiyan torasik displazi sendromu’nun 22 fenotipinin 12.si olan kalıtsal bir sendromdur. Asfiksiyan torasik displazi sendromu'ndaki ortak bulguların büyük bölümü Beemer-Langer sendromu'nda da saptanmaktadır. Ödemli ve şiş ense derisi, skapula ve klavikula malformasyonları, tam gelişememiş meme uçları, diafragmanın yüksek oluşu, hepatosplenomegali ve assit (ascites), anensefali, hidrosefalus ve hipotalamus hamartoması farklı bulgulardır. Yenidoğan ölümlerinin sık görülmesi önemli farklardan biridir.
Meckel sendromu, birden fazla etkisi olan bir genle ilgili (pleiotropic), otosomal resesif yolla aktarılan, 15 kadar fenotipi bulunan kalıtsal bir sendromdur. Çok sayıda ve karmaşık bulguların varlığı tanıda güçlükler oluşturur. Bazı uzmanlar, Meckel sendromunun 3 temel bulgusu olduğunu savunurlar: (i) Santral sinir sistemi malformasyonları ; (ii) Kistik böbrekler; (iii) Karaciğer patolojileri. Bebeklerin yaşama yetisi çok kısıtlıdır; canlı doğabilenlerin çoğu birkaç hafta içimde yitirilir.