Apert sendromu
| Apert sendromu | |
|---|---|
| Diğer adlar | Akrosefali-sindaktili tip 1[1] |
 | |
| Apert sendromlu el sindaktili ile | |
| Uzmanlık | Medikal genetik |
Apert sendromu (acrocephalosyndactyly), otosomal dominant yolla aktarılan kalıtsal bir sendromdur.[2] FGFR2 geni mutasyonuyla ortaya çıkan sendromlar kümesi üyelerinden biridir.[3] FGFR2 geni, fibroblast growth factor reseptörüdür (embriyonun gelişmesinde önemli görevleri vardır; kan damarlarının ve kemiklerin kimi zararlarının onarımında da etkilidir).[2][4][5]
Cücelik boyutlarına ulaşabilen genel bir fiziksel gelişme geriliği vardır. Kafatası eklemlerinden koronal suturanın enken kapanması (kraniyosinostoz) nedeniyle ön-arka boyutunun çok kısa, ancak kaş çıkıntıları-kafatası tepe noktası arasındaki boyutun çok uzun olduğu, makrosefali izlenimi veren bir kafa biçimi vardır; alın (frontal kemik) yüksek ve geniştir (akrobrakisefali).[6] Kafa kemiklerinde defekt alanları saptanır. Kafa tabanı asimetriktir. Gözler arası açıklık artmıştır (hipertelorizm). Göz çukurları (orbita) sığdır. Üst göz kapağında sarkma (ptozis) olabilir. Orta kulak sorunları (otitis media) ve işitme sorunları vardır. Burun sırtı basık, ucu top gibidir.[5][7][8][9][10]
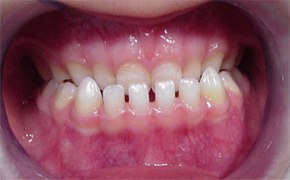
Yüz asimetriktir; yüz orta bölüm hipoplazisi nedeniyle üstçene küçüktür (mikrognati). Üstçene yanlarda dişleri taşıyan kemik dokusu (alveol kretleri) kalın, damak dar ve çukurdur. Üstçene diş arkı V biçimindedir. Sert damak kısadır ve uvula’yı da içine alan bir yarık vardır (yarık damak + uvula bifida).[5][10]
Dişetlerinde büyüme saptanır (gingival hiperplazi). Dişlerin sürmesinde gecikme ve örtüşme bozuklukları (maloklüzyon) görülür. Dil büyüktür (makroglossi). Aşırı bir tükürük salgısı vardır (hipersalivasyon).[8][8][10][11]
Deride aşırı yağlıdır ve bu nedenle çok sayıda akne saptanır. Yüz, gövde ve kollar derisinde içi irinli küçük kabarıklıklar (püstüller) oluşur.[5][9][11]
Sindirim kanalında yemek borusu anomalisi (özofagusu atrezisi; yemek borusunun bir bölümünün konjenital ekskliğidir) ve mide çıkış kapısının darlığı (pilor stenozu) bulguları saptanır. Kalp septum defektleri (atrial; ventriküler) olabilir.[5][8][9][10][11]
Parmak kemiklerinde simetrik sindaktili, kısa parmaklar (brakidaktili) ve eklemlerde kaynaşmalar (ankiloz); omurga sisteminde spina bifida görülür.[5][8][9][10][11]
Erkeklerde skrotuma inmemiş testis, kızlarda vajina atrezisi (vajinanin bir bölümünün konjenitel eksikliği) olabilir. Üriner sistemin en belirgin bulgusu hidronefroz’dur.[5][8][9][10][11]
Çok değişken sayıda beyin anomalileri ve bunlara bağlı zeka geriliği izlenir.[5][8][9][10][11]
Hastalığın progresyon göstermesi halinde solunumda zorluk, işitme kayıpları, görme kayıpları kalıcı hale gelebilir. Bu nedenle zamanında ve yeterli müdahaleler yapılarak komplikasyon gelişiminin önüne geçilmelidir.
Apert sendromunda beklenen ömür ise tamamen hastalığın seyri, komplikasyon gelişip gelişmediği, hastanın çocukluk dönemini atlatıp atlatmadığı ile yakından ilişkilidir.[12]
Kaynakça
- ^ "Apert syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. 18 Mayıs 2019 tarihinde kaynağından arşivlendi. Erişim tarihi: 18 Mayıs 2019.
- ^ a b Slaney SF, Oldridge M, Hurst JA, et al. Differential effects of FGFR2 mutations on syndactyly and cleft palate in Apert syndrome. American Journal of Human Genetics, 58(5): 923–932, 1996
- ^ Bochukova EG, Roscioli T, Hedges DJ, et al. Rare mutations of FGFR2 causing Apert syndrome: identification of the first partial gene deletion, and an Alu element insertion from a new subfamily. Human Mutation, 30:204–211, 2009
- ^ Houssaint E, Blanquet PR, Champion-Arnaud P, et al. Related fibroblast growth factor receptor genes exist in the human genome. Proc. Natl. Acad. Sci. USA (PNAS), 87(20): 8180-8184, 1990
- ^ a b c d e f g h Carinci F, Pezzetti F, Locci P, et al. Apert and Crouzon syndromes: clinical findings, genes, and extracellular matrix. Journal of Craniofacial Surgery, 16(3):361-368, 2005
- ^ Cohen MM Jr, MacLean RE. Craniosynostosis: Diagnosis, Evaluation, and Management, Oxford University Press, NY, 2000
- ^ Kreiborg S, Cohen MM Jr. Is craniofacial morphology in Apert and Crouzon syndromes the same? Acta Odontologica Scandinavica, 56:339-341, 1998
- ^ a b c d e f g Haritha A, Jayakumar A. Syndromes as they relate to periodontal disease. Periodontology 2000, 56:65–86, 2011
- ^ a b c d e f Aydın H, Geçkinli B, Karaman A. Apert sendromlu tipik bir olgu. Göztepe Tıp Dergisi, 29(2):115-117, 2014
- ^ a b c d e f g Maksillofasiyal 14 Haziran 2020 tarihinde Wayback Machine sitesinde arşivlendi. sendromlar, Haziran 2020
- ^ a b c d e f Hohoff A, Joos U, Meyer U, Ehmer U, Stamm T. The spectrum of Apert syndrome: phenotype, particularities in orthodontic treatment, and characteristics of orthognathic surgery. Head & Face Medicine, 3:10-24, 2007
- ^ "Apert Sendromu: Tedavisi ve Yaşam Beklentisi • Doktordan Haberler". doktordanhaberler.com. 5 Eylül 2017. 19 Aralık 2018 tarihinde kaynağından arşivlendi. Erişim tarihi: 20 Şubat 2022.











