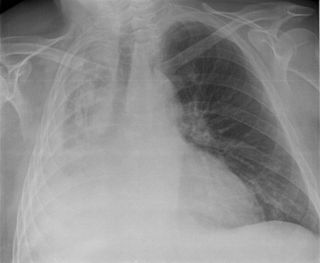
Aort koarktasyonu
Aort darlığı ya da aort koarktasyonu, aort ana arterinin bir bölgede gösterdiği daralmadır. Aortun daralması kan akımının engellenmesine neden olur. Olguların çoğu, yaşamın ilk yılı içindeki bebeklerde saptanan doğumdal (konjenita)) anomalilerdir; bunların bir bölümü de sendroma-özgü bulgular arasında yer alır (örneğin Turner sendromu). Daralma, aortun küçük bir alanını ya da büyükçe bir bölümünü etkiler. Çoğunluğu toraks içindeki aort arkındadır, karın aortundaki daralmalar seyrektir.[1][2][3]
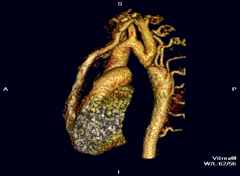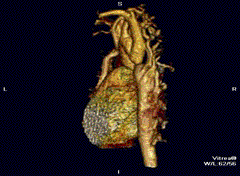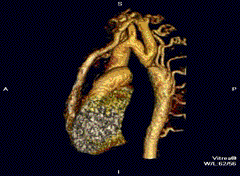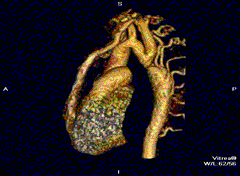
Erkek hastalar çoğunluktadır (Turner sendromunda tam tersi görülür). Kalp-damar sisteminde (kardiyovasküler sistem) tek (izole) anomali olarak saptanan aort darlığı için “basit tip” nitelemesi yapılır. “Kompleks tip” olgularda aort darlığının yanı sıra iki yapraklı aort kapağı (bicuspid aortic valve), ventriküler septal defekt, atrial septal defekt, patent ductus arteriosus, mitral yetmezlik, Willis polgonu anevrizmaları gibi anomaliler de saptanabilir.[4]
Tipleri
İki tip aort koarktasyonu vardır:

(i) Bebek tipi (infantil tip, preduktal tip): Kalpteki aort kapağı ile ductus arteriosus arasındaki daralmalardır. Fetüste, sol kalbe gelen kan akımının azalmasına neden olan bir kalp anomalisinin varlığında ortaya çıkar. Gelen kan miktarının azlığı aortun gelişmesinde aksamaya (hipoplazi) neden olur. Doğumu izleyen ilk aylarda belirti verebilir. Darlık düzeyi arttıkça prognoz da kötüleşir. Patent ductus arteriosus ile birlikte olan olgularda vücudun alt bölümünde morarma (siyanoz) görülür. Bu bebekler ölüm riski yüksektir; herhangi cerrahi bir girişim yapılmazsa doğumu izleyen ilk haftalar içinde kaybedilirler.
(ii) Erişkin tipi (postduktal tip): Ductus arteriosus kapanmıştır. Darlık, aortun toraks bölümündeki arktan çıkan damarlardan sonra (aort arkından çıkan damarların distalinde) oluşur. Daha çok erişkinlerde görülür. Darlığın altında kalan vücut bölümünde arteryel kan basıncı düşük (hipotansiyon), darlık üzerindeki bölümünde ise yüksektir (hipertansiyon).
Aort çapının etkilenmesi farklı düzeylerdedir. Daralma çok az ya da çok fazla olabilir; çok hafif darlıklarda klinik bulgu yoktur. Klinik belirtiler, daralmanın düzeyi kadar patent ductus arteriosus varlığına da bağlıdır. Patent ductus arteriosus bulunmayan olgular erişkinlik dönemine dek belirti vermeyebilir. Klinik bulguların saptandığı bebeklerde beslenme güçlüğü ve solunum sıkıntısı vardır. Bir süre sonra, darlık bölgesinin etkisiyle, daha çok güç kullanmaya çabalayan sol ventrikülde hipertrofi ortaya çıkar. Çocukluk dönemindeki sol ventrikül hipertrofisinde sersemlik, hızlı solunum, yorgunluk atakları, göğüs ağrısı gibi klinik bulgular vardır. Oyun çağı çocuklarında, kesik topallama (claudicatio intermittens) görülür, bacaklar ve ayaklar soğuktur.
Radyoloji
Tanı için görüntüleme sistemlerinin tümünden ekokardiyografi yararlanılır. Daralmış olan aortun iki yakası arasında, darlığın etkilerinin giderilmesi çabası olarak gelişen yardımcı (kollateral) damar ağının varlığı erişkinlerde saptanan tipik bulgulardır. Kostaların radyolojik incelemesinde, çoğalan geniş kan damarlarının basısıyla oluşan kemik aşınmaları saptanır.[5]
Tedavi
Küçük ve komplikasyonsuz darlıklarda metal stent uygulanabilmektedir. Komplikasyonsuz olgularda, dar olan bölge çıkarılır. Büyükçe darlıklara, kalan açıklığa sentetik damar grefti konmaktadır (angioplasti); tüm bu çabalara karşın, çok sayıdaki komplikasyon nedeniylre hastaların yaşam kalitesi düşük, süresi kısadır.[4][6]
Kaynakça
- ^ Kumar V, Abbas AK, Aster JC. Robbins and Cotran Pathologic Basis of Disease. 9th edt., Elsevier Saunders, Philadelphia, 2015
- ^ Anderson RH, Baker EJ, Macartney FJ, Rigby ML (editors). Paediatric Cardiology. Elsevier, Philadelphia, 2002
- ^ Goljan EF. Rapid Review Pathology. 5th edition, Elsevier, Philadelphia, 2019
- ^ a b Yuh D, Vricella L, Yang S, Doty J. Johns Hopkins Textbook of Cardiothoracic Surgery, Second Edition 2nd Edition. McGraw-Hill Medical, New York, 2014
- ^ Kline JS, Brant WE, Helms CA, Vinson EN (editors). Coarctation of the aorta. Fundamentals of Diagnostic Radiology (5th edition; Kindle book), Wolters Kluver, Philadelphia-London-Tokyo, 2019
- ^ Celermajer DS, Greaves K.Survivors of coarctation repair: fixed but not cured. Heart. 88 (2): 113–114, 2002