
Korpus kallozum, beynin her iki hemisferi arasındaki bilgi iletişimini sağlayan sinir ağlarından oluşan yapıdır.
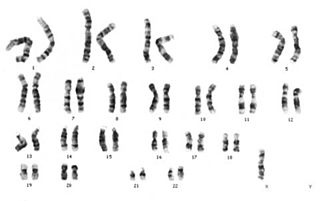
Turner sendromu, kromozom anomalisi sonucu ortaya çıkan ve kız çocuklarını etkileyen bir sendromdur; bir dişide eşey kromozomlarından birinin bulunmaması sonucu ortaya çıkar. Turner sendromluların fenotipi dişi olarak görülür fakat; eşey organları ve eşey hücreleri gelişmez. Kısır bireylerdir.

Klinefelter sendromu ya da 47, XXY sendromu; hücre bölünmesi sırasında, eşeysel kromozom düzensizliklerinden kaynaklanan semptomların kişide görülmesi durumudur.

Frajil X sendromu, X kromozomuyla ilişkili bir zeka (mental) gerilik sendromudur.
Korpus kallozum agenezisi (KKA) ya da nasırsı cisim gelişmezliği ya da yokluğu, nadir görülen bir doğumsal bozukluktur. Korpus kallozum kısmen veya tamamen gelişmemiştir.
Opitz GBBB sendromu 22q11.2 sendromları kümesinin kalıtsal olan tek fenotipidir. 2 tip Opitz GBBB sendromu bilinmektedir.
Ağız-Yüz-Parmak sendromu tip 1 , ektodermal displazi bulguları da içeren, X-kromozomu aracılığıyla dominant (XLD) geçen kalıtsal bir sendromdur. Simpson-Golabi-Behmel sendromu tip 2 ile alelik bağı olduğu belirlenmiştir. Kız çocuklarında görece sıktır. Erkek fetüsler, kalp ve beyin anomalilerinin neden olduğu intauterin ölümler nedeniyle kaybedilirler. Belirgin bir genel gelişme geriliği saptanır.
Holoprosensefali, fetüste, beyin ön bölgesi (prosencephalon) gelişiminin aksaması ve loblarına ayrılamaması olgusudur; ek olarak, mikrosefali, hipofiz bezi ön lobunun agenezi ya da displazisi, yüzde orta çizgi anomalileri, burun malformasyonları, yarık dudak-yarık damak saptanır. 4 tipi vardır:
- Lober tip: Beyin ön lobları oluşmamıştır. En güçlü tiptir.
- Semilober tip: Beyin ön loblarının bir bölümü oluşmuştur, iki hemisfer arasında silik bir çöküntü bulunur.
- Sintelensefali (Syntelencephaly) tipi: Beyin ön lobları hipoplaziktir; ancak parietal loblarla arasında bulunması gereken oluk seçilemez.
- Corpus callosum agenezi içeren tip: Corpus callosum hiç yoktur ya da hipoplaziktir.

Baraitser-Winter sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur; 2 fenotipi vardır. Genel gelişme geriliği izlenir. Kraniyosinostoz nedenli trigonosefali ve mikrosefali saptanır. Boyun kısadır. Hipertelorizm saptanır, kaş çıkıntıları yüksektedir. Göz kapakları iridir ve bilateral ptozis saptanır. Mikroftalmi ile iris ve retina defektleri (koloboma) izlenir. Kulak kepçeleri küçüktür ve aşağıdadır; işitme sorunları vardır. Burun sırtı kalın, ucu büyüktür. Uzun bir filtrum altında ince bir üst dudak bulunur. Alt dudak kalındır. Üst dudak ve damak yarıktır. Altçene geridedir (retrognati). Omurga sisteminde kifoz ve skolyoz türü malformasyonlar saptanır. Başparmak sayısı birden fazladır (polidaktili). Değişik oranlarda konjenital kalp anomalileri ve ürogenital anomaliler görülür. Beyinde corpus callosum anomalileri ile frontal lob anomalisi saptanır; epilepsi ve zeka geriliği bulguları vardır. Genel hipotoninin yanı sıra omuz kaslarında giderek artan güçsüzlük görülür. Deri lenfoması ve lösemi riski yüksektir.
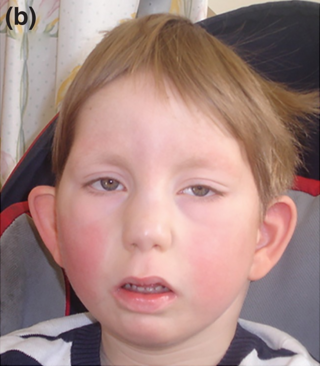
Au-Kline sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur; böbrek, kalp ve yüz bulgularıyla öne çıkar.

Kabuki sendromu, iki fenotipi olan bir sendromdur: Kabuki sendromu 1 ve Kabuki sendromu 2 (Kabuk2). Kabuk1, otosomal dominant yolla aktarılan kalıtsal ya da izole olgular biçiminde görülür; Kabuk2 ise, X-kromozomu aracılığıyla dominant yolla (XLD) aktarılır. Her iki fenotipini çok sayıda ortak bulguları vardır; özellikle yüz, iskelet sistemi, gelişme geriliği, parmakizleriyle ilgili bulgular ve zeka geriliği önemlidir.

Fryns sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Fetüsün amnion sıvısı fazladır (polihidramnios) ve bebek iridir (makrosomi); yenidoğan ölümleri sıktır.

Mowat-Wilson sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Goldberg-Shprintzen sendromu ile çok sayıda ortak bulgusu vardır. Bunlar arasında mikrosefali, psikomotor gerilik, hipotoni, zeka geriliği ve epilepsi en önemlileridir.

Pena-Shokeir sendromu II, otosomal resesif yolla aktarılan, serebrookülofasiyoskeletal sendrom grubundan, kalıtsal bir sendromdur. Cockayne sendromu tip II'nin fenotipi olabilir. Mikrosefali, zeka geriliği, garip yüz yapısı, doğumsal katarakt ve artrogripozis bulgularının ön planda olduğu bir tablodur. İlk 2 yaş içinde yüksek ölüm riski vardır.

Schinzel acrocallosal sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Joubert sendromunun önemli fenotiplerindendir. Santral sinir sisteminde corpus callosum yetersizliği ve zeka geriliği bulguları ile parmaklarda belirgin olan çok sayıda oluşum kusurları saptanır.

Hartsfield sendromu, otosomal dominant yolla aktarılan, ender görülen, kalıtsal bir sendromdur. El ve ayak parmaklarının konjenital eksikliği (ektrodaktili), yarık dudak ve yarık damak ile kafatası malformasyonu (holoprosensefali) üçlüsü temel bulgulardır.

Neu-Laxova sendromu, otosomal resesif yolla aktarılan, 2 fenotipi olan kalıtsal bir sendromdur. Fenotip 2 çok ender görülür. Bebeklerin büyük bölümü ölü doğar ya da çok kısa bir süre yaşayabilir.

Peters’ Plus sendromu , otosomal resesif yolla aktarılan, kalıtsal bir sendromdur. Genel gelişme geriliği nedeniyle boy kısadır. Boyun kalın, alın bombesi yüksektir. Yüz derisinde aşırı kıllanma (hipertrikoz) olabilir. Gözler birbirilerinden uzakçadır (hipertelorizm), göz kapaklarının açıklığı dardır. Görme sorunlarına neden olan Peters anomalisi, glokom, katarakt ve retinafs defektler (koloboma) vardır. İşitme sorunları saptanır; kulaklar küçük ve geridedir, kulak deliği önündeki deride çukurlar görülür. Üst dudak kabarıktır. Yarık dudak ve yarık damak saptanır. Altçene küçüktür (mikrognati). Üst yan kesici dişler eksiktir (hipodonti). Dili ağız tabanına bağlayan bağ kısadır (ankyloglossia).

Vici sendromu, psikomotor gelişme geriliğinin yanı sıra göz, dolaşım sistemi ve bağışıklık sistemindeki defektlerle karakterize, otosomal resesif yolla aktarılan kalıtsal bir sendromdur.

Rubinstein-Taybi sendromu, otosomal dominant yolla aktarılan, 2 fenotipi olan kalıtsal bir sendromdur. 16. kromozom üzerindeki(16p13.3 delesyon) CREBBP(CREB bağlayıcı protein) gen defektiyle bu hastalık oluşur.