
Genetik ya da kalıtım bilimi, biyolojinin organizmalardaki kalıtım ve genetik varyasyonu inceleyen bir dalıdır. Türkçeye Almancadan geçen genetik sözcüğü 1831 yılında Yunanca γενετικός - genetikos ("genitif") sözcüğünden türetildi. Bu sözcüğün kökeni ise γένεσις - genesis ("köken") sözcüğüne dayanmaktadır.

Popülasyon genetiği, popülasyonlardaki fertlerin benzerlik ve farklılıklarının kaynaklarını, bunun yanında popülasyonlardaki alel frekansının dağılımlarını ve değişimlerini araştıran bir genetik altdalıdır.

Peroksizom, hemen hemen tüm ökaryotik hücrelerde bulunan bir organeldir. Çok uzun zincirli yağ asitlerinin, dallı zincirli yağ asitlerinin, D amino asitlerinin, poliaminlerin katabolizmasında ve memelilerin beyin ve akciğerlerinin normal fonksiyonu için önem taşıyan bir eterfosfolipid olan plazmalojenlerin biyosentezi için gereklidir. Ayrıca enerji metabolizması için önemli olan pentoz fosfat yolundaki iki enzimin toplam aktivitesinin yaklaşık olarak %10'unu içerir. Peroksizomların, hayvanlardaki izoprenoid veya kolesterol senteziyle ilişkili olup olmadığı tartışılmaktadır. Filizlenen tohumlardaki glioksilat döngüsü ("glioksizom"), yapraklardaki fotosolunum, tripanazomatidlerdeki glikoliz ("glikozom") ve bazı mayalardaki metanol veya amin oksidasyonu ile asimilasyonu bilinen diğer peroksizomal işlevlerdir.

Frajil X sendromu, X kromozomuyla ilişkili bir zeka (mental) gerilik sendromudur.
Huntington hastalığı (HH), genetik, nörodejeneratif bir beyin hastalığıdır. Hastalarda bazı hareket bozukluklarının yanı sıra mental gerilik görülür. Adını 1872 yılında hastalığın kalıtsal olduğunu ilk olarak gözlemleyen Dr. George Huntington'dan alır. Otozomal dominant olarak kalıtılan bir hastalık olup beyin ve sinir sistemini etkiler. Hasta kişiler genellikle heterozigotlardır. Hastalığın ilk belirtileri 30-50 yaş arasında gözlenir. Hasta bu dönem zarfında çocuk yapmış ise çocuklarına bu hastalığı aktarma riski %50 oranındadır. Hastalık ölümcül olmasa da bu durum hastalığın gelişimine bağlıdır. Hastalığın nedeni Huntington proteinin üretim bozukluğundan dolayı ortaya çıkmaktadır.
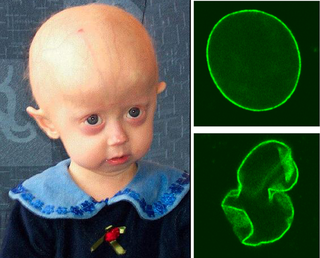
Progeria, halk dilindeki adıyla erken yaşlanma hastalığı, konu hakkında yapılan bilimsel araştırmalar hastalığın çaresini bulmaktan ziyade hastalığa sebep olan faktörleri bulmak ve bu sayede insanlığın ömrünü uzatabilmektir.

National Center for Biotechnology Information (NCBI) United States National Library of Medicine (NLM) ve National Institutes of Health'ın bir bölümü. NCBI 1988 yılında Bethesda'da kurulumuştur.
Rett sendromu, yaygın gelişimsel bozukluklardan birisi olarak sınıflandırılan beyinsel gelişim bozukluğudur. Ancak bunun yanlış bir sınıflandırma olduğunu ve benzer şekilde otistik belirtiler gösteren frajil X sendromu, tüberoz skleroz ya da Down sendromunun yaygın gelişimsel bozukluklar olarak sınıflandırılabileceğini önesüren görüşler bulunmaktadır. Bu sendromun belirtileri kolaylıkla otizm ve Angelman sendromunun belirtileriyle karışır. Klinik belirtiler arasında baş büyüme hızının azalması ve bazen mikrosefali, küçük el ve ayaklar bulunur. Stereotipik ve yineleyici el hareketleri de gözlenir. Bilişsel bozukluk ve gerileme döneminde de sosyalleşme sorunları da belirtiler arasında görülür. Okula girdikleri dönemde sosyalleşme genellikle düzelir. Rett sendromu olan kız çocuklar gastrointestinal bozukluklara yakalanmaya yatkındır ve %80’i nöbet geçirir. Hemen hemen hiç sözel becerileri yoktur ve kadınların %50’si yürüyemez. Skolyoz, büyüme eksikliği ve kabızlık çok yaygındır ve sorunlu olabilir.
Angelman sendromu ilk olarak 1965 yılında İngiliz doktor Harry Angelman tarafından tanımlanmış nörogenetik bir bozukluktur. Irklarda görülme hızı çok iyi bilinmemekle beraber yaklaşık ensidansın 15,000 ila 30,000 canlı doğumda bir olduğu kabul edilmektedir. Anneden gelen kromozom 15'teki bir bozukluktan kaynaklandığı sanılmaktadır. Hastalığın temel bulguları zeka geriliği, yürüyüş-koordinasyon bozukluğu, konuşma bozukluğu, konvülsiyon ve uygunsuz gülümsemelerdir. Hatta bu sebeple hastalık bazen “mutlu kukla ” sendromu olarak da bilinir.

Avram Hershko İsrailli hekim ve biyokimyacı.

Gen düzenleyici ağ veya genetik düzenleyici ağ (GRN), bir hücre içinde (RNA ve protein ifadesinin ürünleri yoluyla) dolaylı olarak birbirleri ile etkileşim gösteren DNA bölümlerinin ve hücre içindeki diğer maddelerin bir koleksiyonu veya derlemesi olup ağlardaki hangi genlerin transkripsiyon sonucu mRNA'ya dönüştürüleceğini belirler. Genel olarak, her mRNA molekülü, belirli bir protein (veya protein dizilerinin) oluşturmak için yola çıkar. Bazı durumlarda bu protein, yapısal proteine dönüşür ve belirli yapısal özellikler oluşturmak için hücre zarında veya hücre içerisinde birikmeye başlar. Başka durumlarda protein bir enzim olacaktır ve bir besin kaynağını veya toksini parçalayarak yıkıma uğratmak gibi belirli bir tepki için kataliz görevini gören bir mikro-makineye dönüşecektir. Bazı proteinler ise sadece diğer genleri uyararak onları aktive etmek görevine sahip olup transkripsiyon faktörleri olarak düzenleyici ağlarda veya ardışık bağlantılardaki ana aktivatörlerdir. Diğer genlerin başlangıç uçlarındaki promotör bölgeye bağlanma ve böylece başka bir proteinin üretimini başlatma suretiyle onları etkinleştirirler ve bu, bu şekilde devam eder. Bazı transkripsiyon faktörleri önleyici olup inhibitör'dür.
Hunter sendromu ya da Tip II mukopolisakkaridoz, iduronat-2-sülfataz (I2S) enziminin eksikliğinden ya da yokluğundan kaynaklanan lizozomal depo hastalığıdır. Doktor Charles A. Hunter (1873-1955) tarafından ilk kez 1917 yılında tanımlandığından, Hunter sendromu olarak adlandırılır.

Şiddetli akut solunum yolu sendromu, insanları etkileyen, şiddetli akut solunum yolu sendromu koronavirüsünün (SARS-CoV) neden olduğu solunum yolu sendromu. Kasım 2002 ve Temmuz 2003 tarihleri arasında Hong Kong'da başlayan SARS salgını neredeyse pandemi hâline gelmiş ve dünya çapında 8422 vaka ve 916 ölüm görülmüştür. Dünya Sağlık Örgütü ölüm oranını %10,9 olarak açıklamıştır. Haftalar içinde SARS erkeni 2003 yılının başlarında Hong Kong'dan 37 ülkeye yayılmıştır.
Alpers hastalığı veya Alpers sendromu çoğunlukla bebek ve çocuklarda görülen merkezi sinir sisteminin ilerleyici dejeneratif bir hastalığıdır. Otozomal resesif bir hastalıktır bu nedenle aktif hastalık için kusurlu genin iki kopyası gereklidir. Tek bir kopya taşıyıcılık anlamına gelir. Alpers hastalığı POLG genindeki bazı genetik mutasyonlar nedeniyle oluşur.
Leigh hastalığı ya da juvenil subakut nekrotizan ensefalomyelopati, Leigh sendromu, infantil subakut nekrotizan ensefalomyelopati ve subakut nekrotizan ensefalomyelopati (SNEM), merkezi sinir sistemini etkileyen nadir kalıtsal nörometabolik ve mitokondriyal bir hastalıktır. İngiliz nöropsikiyatrist Archibald Denis Leigh (1915-1998) tarafından ilk kez 1951 yılında tarif edilmiştir.
Krabbe hastalığı, sinir sisteminin miyelin kılıfını etkileyen nadir, çoğu zaman öldürücü dejeneratif bir hastalıktır. Hastalık sfingolipitlerin metabolizma bozukluğu olan sfingolipidozun bir şeklidir. Kalıtsal olarak otozomal resesif aktarılır. Hastalık Danimarkalı nörolog Knud Haraldsen Krabbe'den (1885-1965) ismini almıştır.
Hermansky-Pudlak sendromu, otosomal resessif aktarılan kalıtsal bir tablodur. 10 fenotipi vardır.

Genetik hastalıklar , bir ailede kuşaktan kuşağa aktarılabilen patolojileri niteleyen tanımlamadır. Kalıtsal hastalıkların gelecek kuşaklara aktarılmasında etkili olan faktörlerler, genlerdeki ve kromozomlardaki yapısal değişikliklerdir.
Gerstmann-Sträussler-Scheinker sendromu (GSS), 20 ila 60 yaş arasındaki hastaları etkileyen, oldukça nadir görülen, genellikle ailesel, ölümcül nörodejeneratif bir hastalıktır. Özel olarak kalıtsaldır ve tüm dünyada yalnızca birkaç ailede bulunur. Bununla birlikte, insan prion proteini olan PRNP'nin oynadığı nedensel rol nedeniyle bulaşıcı süngerimsi ensefalopatiler (TSE) ile sınıflandırılmıştır. GSS ilk olarak 1936'da Avusturyalı doktorlar Josef Gerstmann, Ernst Sträussler ve Ilya Scheinker tarafından rapor edildi.

Ruam atlarda, katırlarda ve eşeklerde görülen zoonotik bulaşıcı bir hastalıktır. Köpekler, kediler, domuzlar, keçiler ve insanlar tarafından da bulaştırılabilir. Burkholderia mallei bakterisi ile oluşan enfeksiyondan kaynaklanır.