De Grouchy sendromu, kromozom anomalisi sonucudur. Büyüme Hormonu eksikliğine bağlı gelişme geriliği saptanır. Baş-boyun bölgesi bulguları önemlidir; kısa bir boyun belirlenir. Mikrosefali, Biparietal kısalık vardır. Gözler çekiktir. Hipertelorizm, optik atrofi, strabismus, nistagmus, glokom gözlerle ilgili olan önemli bulgulardır. Burun kısa-belirgindir. Kulak malformasyonlarına işitme sorunları eşlik eder. Yüz orta bölümünde hipoplazi belirlenir. Filtrum kısa, üst dudak ince ve alt dudak kalıncadır. Üstçenede hipoplazi, altçenede prognatizm vardır. Yanaklar dolgundur. Dudak, damak ve uvula yarıkları saptanır. Kardiyovasküler anomaliler ile defektler ile bunların yol açtığı konjestif kalp yetmezliği, yineleyen solunum yolları infeksiyonları ile astım bulguları göze çarpar. Karın duvarında herniler, erkeklerde dış genital organ anomalileri, gevşek eklemler, skolyoz, parmak-el-ayak anomalileri, deride atopik ekzema bulguları, IgA yetersizliği saptanabilir. Sinir sisteminde serebellum hipoplazisi ve geniş ventriküller, periferik sinirlerde myelinizasyon kusurları vardır; zeka geriliği, psikomotor gerilik, hipotoni, korea ve epileptik ataklar bu bulguların klinik yansımalarıdır.
Waterman sendromu kalıtsaldır ; hipertrikozis/gingival hiperplazi sendromunun fenotipidir. Kaba yüz yapısı ve yüz derisinde aşırı kıllanma saptanır. Aşırı kıllanma tüm vücutta görülebilir (hipertrikozis). Bu bulgulara eşlik eden ikinci önemli bulgu ise yaygın dişeti büyümeleridir. Ayrıca zeka geriliği ve epileptiform ataklar vardır.

Osteokalsin, başka bir adla kemik gama-karboksiglutamik asit içeren protein (BGLAP), kemik ve dentinde bulunan ve ilk olarak civciv kemiğinde kalsiyum bağlayıcı protein olarak tanımlanan küçük (49-amino-asit) kollajenöz olmayan protein yapılı bir hormonudur.
APECED sendromu, ektodermal displazi bulguları içeren, otosomal dominant ya da otosomal resesif geçen kalıtsal bir sendromdur. İlk belirtiler çocukluk yaşlarında ortaya çıkar. Primer immun yetmezlik sendromlarından biridir.

Ellis-van Creveld sendromu, ektodermal displazi bulguları içeren, otosomal resesif geçen kalıtsal bir sendromdur. Asfiksiyan torasik displazi sendromu'nun 22 fenotipinden 2'si Ellis-van Creveld sendromu olarak değerlendirilir.
ADULT sendromu (acro-dermo-ungual-lacrimal-tooth), ektodermal displazi bulguları içeren, TP63 gen mutasyonu sonucu ortaya çıkan, otosomal dominant geçen kalıtsal bir sendromdur.

Coffin-Siris sendromu, ektodermal displazi bulgularını da içeren, otosomal dominant geçen kalıtsal bir sendromdur. 8 fenotipi vardır. Çene-yüz bulguları fenotiplerden 3'ünde belirgindir. Hastalarda genel gelişme geriliği vardır. Kafatası küçüktür (mikrosefali).
Rothmund-Thompson sendromu, ektodermal displazi bulguları da içeren, otosomal dominant geçen, 2 evreli, kalıtsal bir sendromdur.
- Akut evre: 3-6. aylarda başlar. Yüz ve yanaklarda eritemli plaklar belirir, sonraları ekstremitelerde de oluşur. Gövde ve karın derisi etkilenmez.
- Kronik evre: Gelişme geriliğinin saptandığı, yıllarca sürebilen bir evredir. Vücudun farklı yerlerinde, birbirleriyle ilintisiz çok sayıda bulgu saptanır; ektodermal displazi kapsamına giren diş ve deri bulguları oldukça belirgindir:
Johanson-Blizzard sendromu, ektodermal displazi bulguları da içeren, otosomal resesif geçen kalıtsal bir sendromdur. Genel bir gelişme geriliğinin yanı sıra psikomotor gerilik de saptanır.
Konjenital kızamıkçık sendromu , gebelerdeki kızamıkçık (rubella) infeksiyonunun bebeklerde oluşan komplikasyonudur. Gebelikten 1 ay öncesinde ve gebeliğin ilk 3 ayında geçirilen rubella infeksiyonunda risk yüksektir; gebelik süresinin 3. ayı geçtiği olgularda geçirilen infeksiyonlarda risk giderek azalır. Klasik ve bunlara ek olarak saptanabilen teratojen bulgular şunlardır:

Jaffe-Campanacci sendromu, gen mutasyonuyla ortaya çıkan izole sendromlardandır. Deride café-au-lait pigmentasyonu, uzun kemiklerde non-ossifying fibromalar ile bunların neden olduğu patolojik kırıklar, çenelerde dev hücreli reparatif granülom özelliği taşıyan lezyonlar, hipogonadizm ve zeka geriliği bulgularını içerir. Kalıtsal olgular az sayıdadır ve Nörofibromatoz tip 1 grubu içinde yer alırlar.
Multipl endokrin neoplazi (MEN), endokrin bezlerden kökenli çok sayıda tümörlerinin saptandığı, otosomal dominant yolla aktarılan kalıtsal bir sendrom kümesidir. Endokrin tümörlerin yanı sıra, endokrin nitelik taşımayan organlardan ve dokulardan kökenli tümörler de görülmektedir; endokrin kökenli olsun ya da olmasın, iyi huylu ya da kötü huylu (kanser) tümör özelliklerini taşırlar.

Cantu sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur.
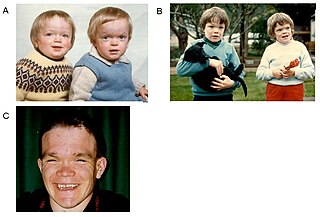
Mannosidozis (mannosidosis), otosomal resesif yolla aktarılan, mannosidaze enzimi eksikliğiyle karakterize kalıtsal bir lizozomal depo hastalığıdır. Alfa-B ve Beta-A olarak nitelendirilen 2 tipi vardır. Alfa-B tipi olgulardaki bulgular çok sayıdadır ve daha güçlüdür.
Aşırı büyüme sendromları , çocuklarda, doğum öncesi (prenatal) dönemde ya da doğumdan sonra (postnatal) dönemde ortaya çıkabilen, bebeklerin/çocukların vücutlarının tümünde ya da bir bölümünde ortaya çıkan irileşmeyle karakterize olgulardır; aşırı büyümelerin görüldüğü sendromlarda, kilo ve boy artışı, çeşitli anomaliler ile zeka geriliği ve tümör riskinin varlığı en sık rastlanan bulgulardır.
Winchester sendromu, MMP14 genindeki spontan mutasyona bağlı ender görülen bir sendromdur.

Zimmermann-Laband sendromu, bir bölümü sporadik, kalan bölümü otosomal dominant yolla aktarılan, dişeti büyümelerinin baskın olduğu ağız bulguları, burun ve kulak kıkırdaklarında yapısal bozukluklar, aşırı kıllanma, omurga eğrilikleri, parmaklarda ve tırnaklarda gelişme aksamaları, karaciğer ve dalak büyümesi (hepatosplenomegali) bulgularının saptandığı, 3 fenotipi olan, otosomal dominant yolla aktarılan kalıtsal bir sendromdur; 2. ve 3. fenotip çok ender görülür.

Kallmann sendromu, gonadotropik hormonun (GnRH) doğumsal yetersizliği sonucu ortaya çıkan gonadortopinlerin yetmezliğine bağlı cinsel olgunlaşma bozukluklarının saptandığı, hipogonadizm olgularının konjenital türü olan "hipogonadotropik hipogonadizm"ler grubunun üyesidir. 25 fenotipi olan konjenital hipogonadotropik hipogonadizmin 1. ve 2. fenotipleri özel nitelik taşırlar; birinci fenotip Kallmann sendromu 1 ve ikinci fenotip Kallmann sendromu 2 olarak tanınmaktadır. Benzer bulgular, hipogonadotropik hipogonadizm dışındaki bazı sendromlarda ikincil bulgulardan biri olarak belirlenir. Kallmann sendromu 1, X-kromozomu aracılığıyla resesif (XLR); Kallmann sendromu 2, otosomal dominant (OD) yolla aktarılır.
IMAGe sendromu, genel gelişme geriliği, genital anomaliler ve konjenital adrenal hipoplazisi ile karakterize, otosomal dominant yolla aktarılan kalıtsal bir sendromdur; otosomal resesif olgular da bilinmektedir.

Konjenital hiperinsülinizm, yenidoğanlarda aşırı insülin üretimi nedeniyle şiddetli hipoglisemiye neden olan nadir bir hastalıktır. HI'nin çeşitli nedenleri vardır ve bunlardan bazılarının genetik mutasyonlar sonucu ortaya çıktığı bilinmektedir. HI bazen tek başına (izole) ortaya çıkar ve nadiren diğer tıbbi durumlarla (sendrom) ilişkilidir.
