Noonan sendromu kalıtsal bir sendromdur; bazı uzmanlarca Turner sendromu'nun erkek çocuklarında görülen tipi olarak tanımlanır. 10 fenotipi vardır, bunlardan fenotip 2 otosomal resessif, öteki 9 fenotip otosomal dominant kalıtım niteliği taşır. Hem çocukları hem de yetişkinleri etkiliyen bir hastalıktır. Genellikle genetik kalp rahatsızlıkları ve iskelet bozuklukları ile ilişkilendirilmektedir.
Diamond-Blackfan anemisi, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Bu sendrom, iki sendromdan oluşan bir grubun ikincisidir. İlk bulgulardan biri olan normokromik-makrositik anemi, bebek 3 aylıkken saptanır. Myelodisplastik sendromla birlikte belirgin bir retikülositopeni tablosu gelişir. Lösemi ve osteosarkom riski yüksektir.
Femoral-facial sendrom, izole sendromdur; diabetik annelerin bebeklerinde görece sık rastlanır. otosomal dominant yolla aktatılmış az sayıda olgu bildirilmiştir.

Fryns sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Fetüsün amnion sıvısı fazladır (polihidramnios) ve bebek iridir (makrosomi); yenidoğan ölümleri sıktır.
Ritscher-Schinzel sendromu, 2 fenotipi olan kalıtsal bir sendromdur.

Mandibulofacial dysostosis sendromu, yüz bulgularının ön planda olduğu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. 1. ve 2. farengeal arklardaki gelişme bozukluğunun sonucudur. "Mikrosefali içeren tipi " ve "Alopesi" içeren tipi önemlidir. “Bauru tipi” çok enderdir. “Ptozis" içeren tip ise kaynaklarda bildirilmiş tek olgu olarak görülmektedir. Treacher Collins sendromu ile yakınlığı tartışılmaktadır.

Bohring-Opitz sendromu, otosomal dominant yolla aktarılan, gelişme ve zeka geriliği bulgularının ön planda olduğu, hastaların çoğunun çocukluk yaşlarında kaybedildiği bir sendromdur. C sendromu'nun fenotipi olarak benimsenir.
Delleman sendromu, spontan gen mutasyonu sonucu ortaya çıkan izole olgulardır.

Genitopatellar sendrom, otosomal dominant yolla aktarılan, kalıtsal bir sendromdur. Mikrosefali ve mikrognati vardır. Saçlar seyrektir. Çekik ve iri gözler, iri bir burun içeren kaba yüz yapısı saptanır. İşitme sorunlarının yanı sıra yarık damak ve dişlerin sürmesinde aksamaları olduğu görülür.

Kapur-Toriello sendromu, otosomal resesif yolla aktarılan, ender görülen, kalıtsal bir sendromdur. Skafosefali olabilir, boyun kısadır. Ensedeki saç çizgisi oldukça aşağıdadır. Garip bir yüz yapısı vardır. Gözler küçüktür (mikroftalmi), göz kapağı düşüklüğü (ptozis) ile katarakt saptanır. Kulak malformasyonları ve işitme sorunları vardır. Burun ucu belirgindir, burun deliklerinden geçen çizgi (columella) uzundur.

Lenfödem-distikiazis sendromu (lymphedema-distichiasis), otosomal dominant yolla aktarılan, ender görülen, kalıtsal bir sendromdur. Pubertede başlayan, bacaklarda saptanan lenf damarları hiperplazisi ve lenfödem tipik bir bulgudur; tabloya lenfanjit eklenebilir. Gözlerde, Meibomian bezlerinin kanallarında kirpikler saptanır; kirpikler iki sıralıdır (distichiasis). Kornea ülserleri, konjunktivit ve yineleyen arpacıklar gözlenir. Göz kapağı düşüklüğü olabilir (ptozis). Ağız incelemelerinde yarık dudak ve yarık damak belirlenir. Kalpte ventriküler septal defekt, Fallot tetralojisi ile aritmiler saptanır. Varisler görülebilir. Bazı hastalarda böbrek etkilenmesi ve diabet bulguları vardır.

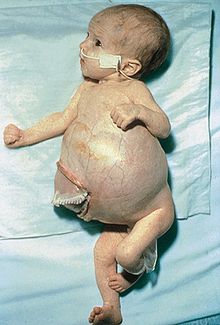
Meckel-Gruber sendromu, çok sayıda genin etkilenmesinden kaynaklanan, bu nedenle 15 fenotipi olan, otosomal resesif yolla aktarılan, siliyopatiler grubundan, kalıtsal bir sendromdur. Kistik böbrekler, beyin anomalileri ve ensefalosel, polidaktili ve karaciğer anomalileri başlıca bulgulardır. En sık görülen fenotip Meckel sendromu 1, öteki fenotiplerdeki bulguların çok büyük bir bölümünü içerir. Bazı fenotipleri Joubert sendromuyla çakışan nitelikleri içermektedir.

Neu-Laxova sendromu, otosomal resesif yolla aktarılan, 2 fenotipi olan kalıtsal bir sendromdur. Fenotip 2 çok ender görülür. Bebeklerin büyük bölümü ölü doğar ya da çok kısa bir süre yaşayabilir.

Peters’ Plus sendromu , otosomal resesif yolla aktarılan, kalıtsal bir sendromdur. Genel gelişme geriliği nedeniyle boy kısadır. Boyun kalın, alın bombesi yüksektir. Yüz derisinde aşırı kıllanma (hipertrikoz) olabilir. Gözler birbirilerinden uzakçadır (hipertelorizm), göz kapaklarının açıklığı dardır. Görme sorunlarına neden olan Peters anomalisi, glokom, katarakt ve retinafs defektler (koloboma) vardır. İşitme sorunları saptanır; kulaklar küçük ve geridedir, kulak deliği önündeki deride çukurlar görülür. Üst dudak kabarıktır. Yarık dudak ve yarık damak saptanır. Altçene küçüktür (mikrognati). Üst yan kesici dişler eksiktir (hipodonti). Dili ağız tabanına bağlayan bağ kısadır (ankyloglossia).
Psödotrisomi 13 sendromu (holoprosencephaly-polydactyly sendromu), otosomal dominant yolla aktarılan, çok sayıda konjenital anomali içeren kalıtsal bir sendromdur. Az da olsa, otosomal resesif olgulara da rastlanabilir.

Oculofaciocardiodental sendrom, göz malformasyonları, çene-yüz anomalileri, dolaşım sistemi ve bağışıklık sistemi defektleriyle karakterize, X-kromozomu aracılığıyla dominant (XLD) yolla aktarılan kalıtsal bir sendromdur. Mikroftalmi sendromunun 18 fenotipinden biridir.
Hydrolethalus sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Bebeğin ölü doğumuna ya da doğumdan sonraki ilk birkaç saat içinde ölümüne neden olan çok sayıda malformasyon saptanır. Farklı genlerin etkilendiği 2 fenotipi vardır. Finlandiyalılarda sık görülür.

Konjenital kalp defektleri ya da konjenital kalp hastalıkları, kalp işlevlerini olumsuz yönde etkileyen, sık görülen doğumsal patolojilerdir. Gebeliğin 3.-8. haftalarındaki embriyogenez kusurunun sonucudur. Doğumların %1’ında görülürler; prematürelerde ve ölü doğum bebeklerinde daha sıktır.

Rubinstein-Taybi sendromu, otosomal dominant yolla aktarılan, 2 fenotipi olan kalıtsal bir sendromdur. 16. kromozom üzerindeki(16p13.3 delesyon) CREBBP(CREB bağlayıcı protein) gen defektiyle bu hastalık oluşur.
Meckel sendromu, birden fazla etkisi olan bir genle ilgili (pleiotropic), otosomal resesif yolla aktarılan, 15 kadar fenotipi bulunan kalıtsal bir sendromdur. Çok sayıda ve karmaşık bulguların varlığı tanıda güçlükler oluşturur. Bazı uzmanlar, Meckel sendromunun 3 temel bulgusu olduğunu savunurlar: (i) Santral sinir sistemi malformasyonları ; (ii) Kistik böbrekler; (iii) Karaciğer patolojileri. Bebeklerin yaşama yetisi çok kısıtlıdır; canlı doğabilenlerin çoğu birkaç hafta içimde yitirilir.